Evidence for genetic susceptibility to coronary heart disease
Coronary heart disease is a complex phenotype which arises from the interaction of a number of risk factors including smoking, hyperlipidemia, hypertension, obesity, and diabetes. Epidemiologic studies have consistently indicated the importance of a family history of coronary heart disease (CHD) as an independent risk factor for disease. Perhaps the most frequently cited study in this regard is the 1994 report from the Swedish Twin Study.1 That report included 21 004 individuals, of whom 2810 had fatal CHD. Among male twin pairs in which the first twin had died of CHD before the age of 55, the relative risk of fatal CHD in the second twin was 8.1 (95% confidence interval (CI) 2.7–24.5) for monozygotic (i.e. genetically identical) twins and 3.8 (95% CI 1.4–10.5) for dizygotic twins. Among female twin pairs in which the first twin had died of CHD before the age of 65, the relative risk of fatal CHD in the second twin was 15.0 (95% CI 7.1–31.9) for monozygotic twins and 2.6 (95% CI 1.0–7.1) for dizygotic twins. These results clearly indicate a significant genetic contribution to the risk of CHD death. A 2002 analysis of the same cohort, which at that time included 4007 CHD deaths and a follow-up time of up to 36 years, used more sophisticated statistical modeling approaches to conclude that the heritability of fatal CHD events was 57% for males and 38% for females.2 By contrast with the earlier report from this study, it was suggested that there was persistence of the excess genetic risk of CHD death even into old age. A similar analysis among 7955 Danish twin pairs including 2476 CHD deaths reported a heritability of fatal CHD events of 53% for males and 58% for females, which is broadly concordant with the Swedish data.3 Studies in the offspring of the original Framingham Heart Study participants have shown strong evidence for association of parental cardiovascular disease with the risk of offspring cardiovascular disease, and for sibling–sibling association of cardiovascular disease even after multivariable adjustment for other measured risk factors. In these studies, the relative risk associated with parental or sibling cardiovascular disease was between 1.45 and 2.0.4,5
The INTERHEART case–control study identified nine modifiable risk factors (smoking, dyslipidemia, hypertension, diabetes, abdominal obesity, psychosocial factors, daily consumption of fruit and vegetables, regular alcohol consumption, and regular physical activity) which together predicted a very substantial proportion of the risk of a first myocardial infarction (MI) in populations derived from every inhabited continent.6 Family history was also assessed as a risk factor in that study. After adjustment for age, sex, smoking and region of recruitment, a positive family history conferred an odds ratio (OR) for MI of 1.55 (95% CI 1.44–1.67). After additional adjustment for all of the nine most significant factors, the risk was reduced to 1.45 (95% CI 1.31–1.60), remaining significant.
Some studies have examined the heritability of quantitative phenotypes related to atherosclerotic disease, such as carotid intima-media thickness and arterial calcification in various sites;7–9 substantial heritabilities have been found for these phenotypes (30–60%). These findings further substantiate the assertion that genes play a significant role in susceptibility to atherosclerosis and its consequences.
Genetic architecture of coronary heart disease susceptibility
While it is clear that there is a substantial genetic component to CHD susceptibility, the architecture of that component–in terms of the number of effects and their sizes–remains uncertain. Population genetic theory would predict that the size of the effect of an allele on a phenotype should vary inversely with its frequency: that is, alleles that are common in the population should have small biologic effects, while large effects should be confined to rare alleles. With relatively few exceptions, this prediction has been found to hold true both in humans and in experimental animals. Figure 21.1 illustrates this concept. Most of the successes in CHD genetics before 2007 were confined to the upper left-hand portion of the curve shown in Figure 21.1, where studies in families with Mendelian (single-gene) forms of CHD had revealed large-displacement alleles which substantially affected the function of particular genes and which were sufficient alone to confer very markedly increased risks of CHD (for example, LDL-receptor mutations which cause familial hypercholesterolemia). However, because these alleles are rare, each one possibly existing in only a handful of families worldwide, their individual contribution to the population burden of disease is modest. Common alleles which had only small effects on disease risk (conferring odds ratios of 1.2–1.5) would contribute far more significantly to the impact of disease in the population. Conversely, however, such alleles would not be expected to have great predictive value for individual patients.
Figure 21.1 Effect size and allele frequency in populations. Both population genetic theory and empiric evidence suggest that the frequency of an allele will be inversely related to the size of its biologic effect (curve). Three general classes of detectable variants (boxes) can be distinguished; different approaches are needed to detect members of the different classes. GWA, genome-wide association.
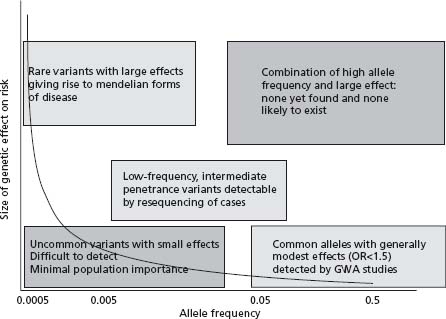
CHD is, in evolutionary terms, a very recent disease. Moreover, even at the peak of the CHD epidemic that is now waning in most Western countries, deaths from CHD tended to occur at ages older than the best estimates of the average life expectancy throughout human history.10,11 Therefore CHD, in common with many other degenerative diseases of later life, has almost certainly been invisible to natural selection throughout our evolutionary history, as it would not have affected our ancestors’ reproductive fitness. This implies that common variants (which tend to be evolutionarily ancient) with effects on CHD could have arisen and persisted by chance alone, although the effects of such variants would be expected to be small. It is an attractive hypothesis that variants with beneficial effects on conditions of greater evolutionary impact (for example, infectious disease or the ability to survive trauma) could have been selected for in our evolutionary history but have now “outlived their usefulness” in the toxic environmental setting of caloric abundance and limited physical activity. However, this has yet to be convincingly demonstrated for any variant.
In the last year, studies which have characterized common variation throughout the human genome in large numbers of CHD cases and controls have considerably clarified our understanding of the situation with respect to the contribution of such common variation to disease risk. One locus has been convincingly replicated in multiple studies thus far (even in relatively small studies), and it seems likely that this is the principal susceptibility locus existing in the genome. It is, however, clear that there remains a substantial “heritability gap”–that is, the fraction of the heritability of CHD that is accounted for by the existing confirmed candidate genes (such as variants in the apolipoprotein E gene) and the newly discovered alleles is far below the total calculated heritability of the condition. Among the possible explanations are that the rest of the heritability is due to very small effects of common alleles which lie beyond the resolution of even the largest association studies. Alternatively, the remaining heritability may be due to “low-frequency intermediate penetrance” variants, which would not have been captured by the existing genome-wide association studies but could nonetheless have significant effects on risk for those individuals who carry them. Some evidence in favor of the existence of such variants from systematic resequencing studies is described below; recent technologic innovations which have dramatically increased the speed and cost effectiveness of genome sequencing will enable systematic genome-wide evaluation of rare variants in the near future.
Mendelian disorders associated with coronary artery disease
A number of Mendelian disorders associated with premature CHD have been recognized, as summarized in Table 21.1. Such disorders explain only a very small proportion of CHD cases, but knowledge of them is important for a number of reasons. First, it allows the development of effective strategies for the detection, treatment and prevention of disease in affected individuals and families, which is particularly important as disease in these individuals often presents at a young age and follows an aggressive clinical course. Second, certain genes causing Mendelian forms of CHD have also been implicated in the risk of non-Mendelian CHD in the general population (see Table 21.1). Third, novel insights into the pathophysiologic mechanisms of disease gained through the study of Mendelian disorders may allow the identification of new biomarkers or targets for therapeutic intervention. Insights from investigations of familial hypercholesterolemia were important in the development of statins, and studies of apolipopro-tein A-I (Apo A-I) deficiency have resulted in recent clinical trials of recombinant Apo A-I Milano for the treatment of CHD, as discussed below.
Table 21.1 Mendelian disorders involving coronary artery disease
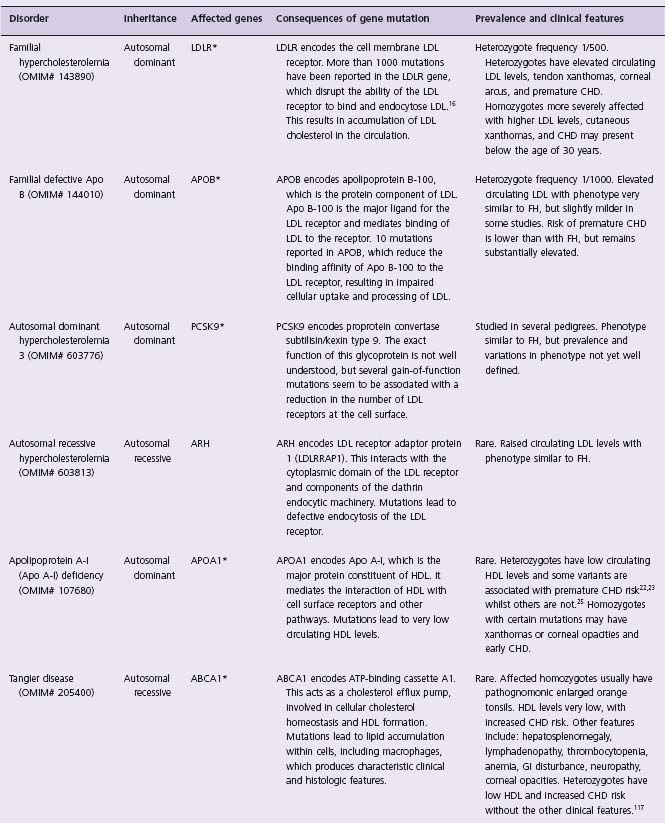
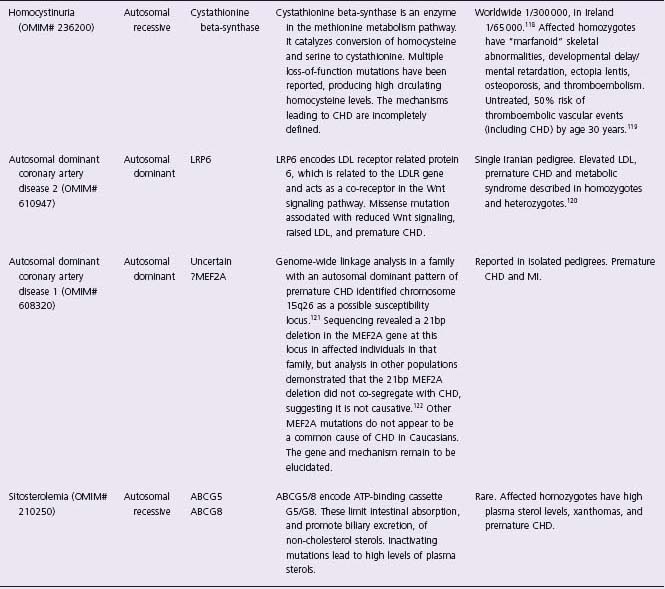
OMIM#, National Center for Biotechnology Information Online Mendelian Inheritance in Man identifier (www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM). *, genes for which variants have been shown to contribute to lipid/cardiovascular phenotypes in the general population. FH, familial hypercholesterolemia.
Familial hypercholesterolemia (FH) is the most common Mendelian disorder conferring CHD risk. It is an autosomal dominant condition caused by mutations in the low-density lipoprotein receptor (LDLR) gene, which disrupt the ability of the receptor to bind LDL at the cell membrane. Cellular uptake of LDL cholesterol is impaired, resulting in increased circulating levels of LDL cholesterol and stimulation of intracellular cholesterol synthesis.12–15 Over 1000 mutations in the LDLR gene have been reported to be associated with the syndrome. Point mutations account for 91% of mutations (missenses, deletions, nonsenses, insertions and splice variants have all been described), and 9% are major rearrangements.16 The heterozygote frequency of familial forms of hypercholesterolemia is around 1 in 500 in Western populations. The term familial hypercholesterolemia is usually used specifically for the syndrome associated with mutations in the LDLR gene, but similar phenotypes are associated with abnormalities of the APOB and PCSK9 genes. Detailed investigation of patients with “familial hypercholesterolemia” phenotypes demonstrated that 79.1% were due to LDLR mutations, with 5.5% due to APOB mutations, and 1.5% due to mutations in PCSK9.17 Involvement of other genes may account for some of the remaining proportion.
FH heterozygotes have elevated circulating LDL cholesterol levels, typically in the range 190–465mg/dL, and develop tendon xanthomas, corneal arcus and premature coronary artery disease often before the age of 50 years. Affected homozygotes are more severely affected, often with LDL cholesterol above 465 mg/dL, cutaneous xanthomas, and coronary disease which may present below the age of 30 years. Untreated, the risk of CHD in affected heterozygotes by age 60 years is approximately 50–85% for men and 30–55% for women.18,19 The key elements of the clinical diagnostic criteria for FH are raised serum cholesterol, tendon xanthomas in the patient or a first-degree relative, and a dominant inheritance pattern of premature CHD or elevated cholesterol.20 Opportunistic and cascade screening for relatives of affected individuals may be used in routine practice, as discussed below.
Apo A-I is the major protein constituent of high-density lipoprotein (HDL). Mutations in the APOA1 gene cause Apo A-I deficiency, which is an autosomal dominant disorder. Numerous mutations in the APOA1 gene have been reported, some of which lead to very low circulating HDL levels and increased risk of atherosclerosis.21–23 However, a variant of Apo A-I (named Apo A-I Milano) identified in an Italian family in 1980 did not seem to be associated with increased CHD risk.24 The pedigree carrying this mutation was traced to a village in northern Italy, revealing 33 heterozygous carriers of the mutation who had markedly reduced HDL (10–30 mg/dL), raised triglycerides but a lower, rather than higher, risk of atherosclerosis compared to the general population.25,26 The Apo A-I Milano protein differs from native Apo A-I by the substitution of cysteine for arginine at position 173, which changes its properties and allows the formation of disulfide-linked homodimers and heterodimers with apoA-II.27 Recombinant Apo A-I Milano complexed with a naturally occurring phospholipid has been manufactured to mimic the properties of nascent HDL, and studies in animal models of atherosclerosis showed that infusions of this complex rapidly reduced the lipid and macrophage content of atherosclerotic plaques.28–31 A randomized double-blinded controlled trial of recombinant Apo A-I Milano therapy on coronary atherosclerosis in human subjects with acute coronary syndromes showed that intravenous infusion of the recombinant Apo A-I Milano complex at weekly intervals for five weeks significantly reduced coronary atheroma volume measured using intravascular ultrasound.32 The mechanism is uncertain, but may relate to an increase in reverse cholesterol transport from atheromatous lesions to the serum with subsequent hepatic removal. These investigations suggest that recombinant Apo A-I Milano, or drugs mimicking the effect of the mutation on lipoprotein trafficking, could be useful for the stabilization, and possibly even regression, of atheromatous plaque in the wider population with CHD.
Family-based studies of non-Mendelian coronary heart disease: genome-wide linkage analysis
Family-based linkage studies, which had delivered the identity of over 2000 genes for Mendelian conditions by 2005, have also been performed in many complex diseases, including CHD. In this approach, the inheritance of chromosomal segments is tracked through families which have multiple members affected with disease by typing markers spaced throughout the genome. Because the individuals involved in a family study are separated by few meioses, comparatively few markers are required to accurately identify the inheritance of each chromosomal segment genome-wide within a family (several hundred rather than the hundreds of thousands of markers required in genome-wide association studies). Segments that are shared more often than expected by chance among family members affected with disease can thus be identified; these segments may harbor genes which are involved in disease etiology. Most such studies in CHD were conceived in the early to mid 1990s, predating the vast expansion (by three orders of magnitude) in knowledge of genetic markers that made genome-wide association studies possible. Such studies were dogged by two intrinsic problems. First, although linkage is a very efficient approach to detect alleles of large effect (such as in the context of Mendelian disease), the power to detect alleles with small effects (odds ratios below about 2.0) is very low.33 Second, even when a replicated linkage “hit” is obtained in a complex disease, the genomic region requiring investigation before a gene is identified remains very large, possibly containing hundreds of genes. As a result, there are relatively few examples where complex disease loci have been identified using this approach.
Several genome-wide linkage studies in CHD have been published,34–40 but cross-study replication of identified loci has in general been lacking. Of these linkage studies, only one produced evidence in favor of a specific positional candidate gene, ALOX5AP, which encodes 5-lipoxygenase activating protein (FLAP).38 FLAP is a necessary co-activator of the enzyme arachidonate 5-lipoxygenase (ALOX5) which synthesizes leukotriene A4 (LTA4), a short-lived mediator of inflammation. Variants in ALOX5AP were associated with a relative risk of MI of 1.8, and a relative risk of stroke of 1.67, in that study. These genetic findings have, however, not been consistently replicated in other studies, and the summary of the evidence to date suggests that the effect of the haplotypes studied, if it exists, is of a much smaller size than originally estimated.41–46 Based on the initial genetic findings, a clinical trial was conducted to examine the effect of FLAP inhibition on levels of biomarkers associated with MI risk in 191 patients who had already suffered an MI and carried at-risk variants in FLAP.47 The FLAP inhibitor led to suppression of plasma levels of leukotriene B4, myeloperoxidase, and C-reactive protein without any adverse events. This result suggests that clinical trials of agents inhibiting FLAP that are powered to detect differences in CHD endpoints would be of interest.
Single nucleotide polymorphisms and the human haplotype map
In the last 18 months genome-wide association (GWA) studies have delivered over a hundred new loci for upwards of 40 common diseases and continuous traits with a “complex” (that is, made up of multiple genetic effects) genetic architecture. The majority of such studies have been conducted in large numbers of unrelated cases of disease and controls, and at its simplest, the analysis consists of comparing the frequency of genotypes at a large number of genetic polymorphisms spaced throughout the human genome one by one in the cases and controls. The polymorphisms used in these analyses consist of single base alternatives in the genetic sequence, called single nucleotide polymorphisms (SNPs, usually pronounced “snips”). The large-scale sequencing studies conducted to complete the Human Genome Project established that SNPs were the most common type of human genetic variant, and that SNPs with a minor allele frequency of greater than 1% occurred on average every 300 bases throughout the human genome.
Consider an autosomal SNP which has as alternative alleles the nucleotides thymine (T) or adenine (A). A diploid individual has three possible genotypes: thymine/thymine (T/T), thymine/adenine (T/A) or adenine/adenine (A/A). If the frequency of these two alleles in the population were 80% T alleles and 20% A alleles, then the three genotype frequencies would be expected to be around 64%, 32%, and 4% respectively (since the frequency of each homozygote genotype would be expected to be the square of the frequency of the corresponding allele, and the heterozygote frequency twice the product of the two allele frequencies). Such a SNP would qualify as a “common variant”–though the cut-off is somewhat arbitrary, SNPs with minor allele frequencies greater than 5% are generally considered to be common. Common SNPs tend to be evolutionarily ancient, because rare variants have a much higher likelihood of being eliminated over time by the random selection of gametes that occurs from generation to generation. Alleles conferring susceptibility to common degenerative diseases of later life such as CHD may have persisted throughout evolution because they do not commonly affect fitness in the reproductive years and are therefore invisible to natural selection. Thus, genetic susceptibility to common disease might arise from a relatively small number of common variants in a particular susceptibility gene, as opposed to a large number of individually rare variants. This theory is known as the “common disease–common variant hypothesis”. It is the main theoretical argument in favor of studying common SNPs for association with disease. From a practical point of view, rare SNPs with small effects on disease would be essentially impossible to detect in studies of a feasible size (see Fig. 21.1), and the technology to systematically investigate genes for rare SNPs, even of large effect, has until very recently been far behind that available for the investigation of common SNPs. Recent GWA studies of many diseases have convincingly shown that the common disease–common variant hypothesis is to an extent true, although even for diseases such as type 2 diabetes, in which a large number of susceptibility variants have been found, these together do not explain the bulk of the inherited propensity to disease.
A systematic discovery program for SNPs was implemented by the SNP Consortium, a public–private partnership formed in 1999. The consortium released 1.4 million SNPs into the public domain by 2001, and there are presently more than 7 million validated SNPs in public databases. This constitutes a fairly complete picture of human common SNP variation. However, simple knowledge of the SNPs did not make the task of assessing variation across the genome possible, since no platform exists that could type 7 million SNPs in each of a large number of cases and controls in a cost-effective manner. It was therefore necessary to additionally determine the relationships between the SNPs that had been discovered, in order to establish which SNPs gave redundant or substantially overlapping information about genetic variation in different regions of the genome, and establish a minimal set of SNPs that could be feasibly typed in case–control studies while retaining relatively full information. Such data were provided by the collaborative International Haplotype Map (HapMap) Project, which aimed to obtain detailed genome-wide information on patterns of common genetic variation in multiple world populations, suitable for association studies (www.hapmap.org).
Chromosomes in the present-day population are essentially mosaics which reflect the results of new mutations and genetic recombination on ancestral haplotypes (Fig. 21.2). When a SNP arises on a chromosome (since the per-base mutation rate at SNPs is low, most SNPs are thought to have arisen only once during evolution), initially the alleles present at all the surrounding pre-existing SNPs are strong predictors of genotype at the new SNP. As meiotic recombination occurs throughout the generations, the initially lengthy chromosomal segment containing strongly predictive genotypes will break down. Note that although in general terms more closely neighboring SNPs will be better predictors of each other’s genotypes, the relationships cannot simply be derived mathematically; they must be empirically observed for each pair of SNPs of interest. The degree to which genotype at one SNP predicts genotype at a neighboring SNP is called “linkage disequilibrium”, abbreviated “LD”. For the purposes of association mapping it is most usefully measured by the correlation coefficient r2 which varies between 0 in the case of two SNPs whose genotypes have no predictive value for each other, and 1 in the case of two SNPs that are perfect proxies for each other (and therefore one of them is entirely redundant for association study purposes). r2 is a useful measure because the power of an association study where the disease-causing variant is not directly observed varies inversely with r2. Thus, if a study of 2000 cases and controls were required to have adequate power to detect an odds ratio of a particular size should the causative variant be directly typed, a study of 4000 would be required if the most closely correlated SNP that was typed had an r2 of 0.5 with the untyped causative SNP.
Figure 21.2 Decay of allelic association over time: chromosomes are mosaics. Consider a new disease susceptibility allele D arising on an ancestral chromosome (bar). In that generation, all the markers in the region including markers M1 and M2 will be correlated with D. Genetic recombination will break down the region of association over time, introducing new haplotypes into the region. If the mosaic is “coarse” (which is the case with the human genome) fewer markers are needed to tag each piece (or “haplotype block”) of the mosaic: thus, M1 and M2 are both still good markers for D when the mosaic is coarse, but only M2 is a good marker after further time has elapsed and the mosaic is finer.
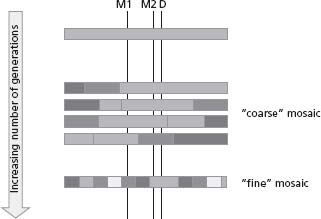
The “grain” of the human genomic mosaic was a key factor in determining the feasibility of GWA studies (Fig. 21.2). If it had been very “fine”, it might have been necessary to type most of the 10 million common SNPs to represent them all satisfactorily, and if relatively “coarse”, typing of only a few hundred thousand SNPs might be sufficient. Some early studies48 provided encouraging information that at the level of single genes, the mosaic was indeed quite coarse; subsequent studies in large genomic segments confirmed that this was more generally true,49 and showed that the mean size of regions of strongly associated SNPs (also known as “haplotype blocks”) is around 22 kb in populations of European or Asian ancestry, and 11 kb in populations of African ancestry.50 These findings, among others, suggested that a substantial proportion of common variation genome-wide could indeed be characterized with several hundred thousand SNPs that were sufficiently well correlated with all the remaining untyped SNPs that they could be considered to “tag” the untyped SNPs. In 2005 the Phase I HapMap reported data from around 1 million SNPs; in 2007, the Phase II HapMap containing data on 3 million SNPs covering most common variations in most of the human genome was made available.51–53 This provided the necessary information to design experiments in which common variation either within candidate genes or genome-wide could be thoroughly assessed using a subset of “tag” SNPs. An excellent recent review provides more detailed information on the achievements of the HapMap.54
The key technologic advance required for the success of genome-wide association studies has been the development of “gene chips” able to assay up to 1 million SNPs chosen on the basis of the genomic knowledge from the HapMap. Currently such chips are available for less than 750 US dollars per sample, and the price is constantly falling even as the genomic coverage improves, in a situation analogous to that for computer microprocessors in recent years. The current “top of the range” products from the major manufacturers Illumina and Affymetrix tag 80-90% of genome-wide SNPs in non-African origin population at an r2 of 0.8 or greater, although only around 66% of genome-wide SNPs are tagged at r2 of 0.8 or greater in African origin populations in whom there is greater haplo-type diversity.
Finally, large samples of well-characterized cases and controls have been required to render both reliable candidate-gene association studies and GWA studies feasible. Early work on CHD genetics in the 1990s clearly showed the potential for false-positive results in small samples; at that stage the problem was one of overoptimism, as investigators hoped to discover genetic effects much larger than those that (with the benefit of hindsight) appear to be present. In the initial stage of a GWA study, where up to 1 million SNPs may be typed, around 50000 SNPs would be expected to show statistical significance at the P < 0.05 level, most of these purely by chance. To avoid these false positives, extremely stringent levels of significance must be adopted (typically 5 × 10-7 or thereabouts). In order to obtain this level of significance for a SNP with a minor allele frequency of around 0.1, that confers an odds ratio of disease of about 1.3, 2000 cases and a similar number of controls are required. Within-study replication of associated SNPs in additional cohorts is considered an additional requirement to rule out the play of chance as a cause for the initial finding.
Candidate-gene association studies in coronary heart disease
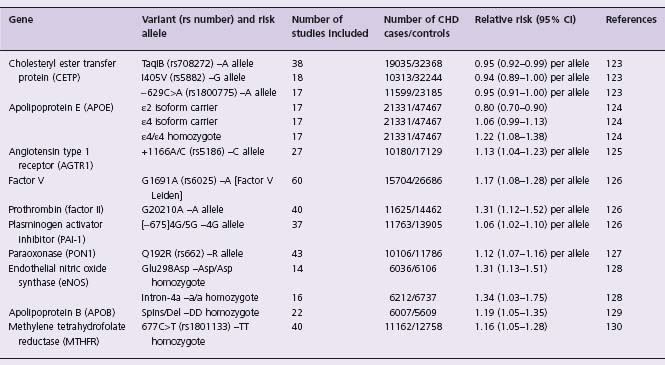
In common with many other diseases, CHD has been the subject of large numbers of candidate-gene association studies that have examined many different genes. Nearly 1000 such published studies are currently in the Genetic Association Database, an archive of association studies in human complex diseases (http://geneticassociationdb.nih.gov). Conclusive identification of associated variants has been very rarely achieved. Several factors are likely to contribute to this. The prior probability of association of any particular SNP in any one of 20000 genes with a given disease is vanishingly small. Relevant pathophysiologic information that has led to the selection of a gene as a candidate (for example, its involvement in determining plasma levels of cholesterol) may increase the prior probability, but even so, it is certain that in this context P < 0.05, the conventionally accepted significance level for result reporting, does not represent secure evidence in favor of the involvement of that gene. Thus, most genetic associations initially reported with significance levels 0.01 < P < 0.05 can reasonably be expected to be false. The majority of reported studies to date have been of insufficient size to robustly identify (i.e. with appropriately small P -values) the effects of small magnitude that would usually be expected for common SNPs. In a 2007 paper, Morgan and colleagues highlighted the replication problems in the field by typing 85 variants in 70 genes previously claimed to be associated with the risk of acute coronary syndrome, finding just one borderline significant (P = 0.03) association that was most likely due to the play of chance.43 Although meta-analyses may, as in traditional epidemiology, assist in identifying true associations, a number of meta-analyses of genetic association studies have shown evidence for publication bias–the preferential reporting in the literature of small positive studies, whereas equally sized negative studies go unreported. This greatly complicates the interpretation of quantitative overviews of the published literature; statistical tests for publication bias are relatively insensitive, and the regular detection of publication bias in the field tends to undermine confidence that it is not present for a particular association even in the absence of statistical evidence. Results of several large-scale (that is, involving greater than 5000 cases of disease) meta-analyses are shown in Table 21.2. Of these, the most convincing evidence in favor of association is with the Apo E epsilon-2/epsilon-3/ epsilon-4 isoform polymorphism. Apolipoprotein E is a ligand for receptors that clear remnants of chylomicrons and very low density lipoproteins; the three isoforms bind with different affinities and are associated with differences in levels of LDL-cholesterol. The recent GWA studies (see below) have not provided a great deal of additional evidence pertinent to this association because the two polymorphisms (Cys112Arg and Arg158Cys) that together define the isoform are not well “tagged” by the SNPs present on the genotyping chips used in studies to date.
Table 21.2 Candidate gene polymorphisms associated with CHD in large-scale meta-analyses (>5000 CHDcases)
The first report from an association study of CHD that typed many thousands of SNPs (although providing insufficient coverage to be classified as a true genome-wide association study) was published in 2002. Ozaki and colleagues examined almost 93000 gene-based SNPs (mainly in exons, introns and flanking regions surrounding genes) in 14 000 genes, in 94 cases of MI and 658 controls.55 Those SNPs showing a P-
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


