Genetics of Channelopathies and Clinical Implications: Introduction
The term channelopathy refers to a genetic cardiac disorder characterized by altered cardiac excitability, in the absence of structural cardiac involvement (with few exceptions). Diseases belonging to this group are also often referred as to inherited arrhythmogenic diseases (IADs). Indeed, growing evidence shows that not only ion channels but also other proteins involved in the control of cardiac excitability are affected.
IADs typically manifest with peculiar electrocardiographic features, syncope, and sudden death in young, otherwise healthy individuals. The common denominator of these disorders is a genetic mutation affecting the genes that control the excitability of myocardial cells (Fig. 36–1).
Figure 36–1
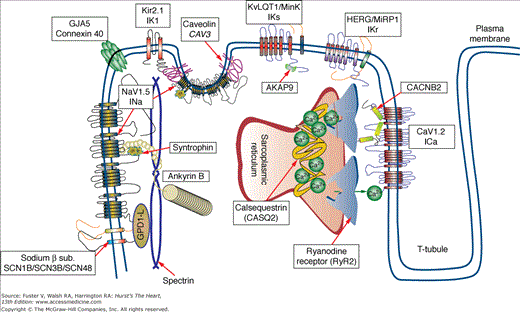
Proteins involved in inherited arrhythmogenic disorders. Diagram showing the intracellular localization of the proteins involved in the pathogenesis of inherited arrhythmogenic syndromes. Each protein can be involved in more than one disease. The figure shows that not only ion channels but also several ion channel interacting proteins are involved in the pathogenesis of inherited arrhythmia syndromes.
The groundbreaking discoveries starting in the 1990s until the beginning of the 2000s gathered the fundamental knowledge on the major genes causing cardiac channelopathies. Stems of such knowledge are the availability of genetic diagnosis, genotype-phenotype correlation, and genotype-based risk stratification schemes that are currently used in clinical practice.
Another interesting area of research that is going to represent an important source of information to link the genetic substrate with the clinics (thus leading to improved risk stratification and clinical management) derives from the evidence of variable expressivity and incomplete penetrance in these disorders. Differential severity of clinical phenotypes is frequently observed not only in patients with mutations on the same gene, but also among patients who harbor exactly the same mutation. Several studies have been recently published that genetic polymorphisms (single nucleotide polymorphisms [SNPs]) may significantly impact cardiac repolarization and the risk of cardiac events. The identification of such modifiers and, even more, the implementation of these findings into clinically applicable tools are in their infancy, but it may be anticipated that this information will be routinely applied for patient management in the near future.
Long QT Syndrome
The long QT syndrome (LQTS) is an IAD in the structurally normal heart characterized by abnormally prolonged QT interval with peculiar morphologic abnormalities of the T wave. LQTS manifests with syncope and/or cardiac arrest typically occurring in children or teenagers. Two major phenotypic variants have been described in the early 1960s: one autosomal dominant (Romano-Ward syndrome) and one autosomal recessive (Jervell and Lange-Nielsen syndrome) also presenting with sensorineural deafness. Additional rare variants including extracardiac involvement are the Andersen syndrome and the Timothy syndrome. The estimated prevalence of LQTS is between 1:7000 and 1:3000, but these should be considered rough estimates because the incomplete penetrance of the disease can lead to underestimation of the actual prevalence (Fig. 36–2).1,2
Figure 36–2

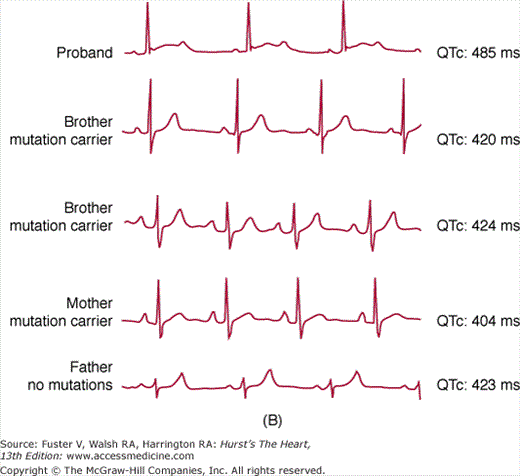
Incomplete penetrance in long QT syndrome (LQTS). Upper panel shows the distribution of QTc interval values in a cohort of 521 genetically affected LQTS patients in comparison with their non–genetically affected family members (n = 594). A remarkable overlap of values is evident in the QTc range between 420 and 460 ms. Lower panel depicts a typical example of incomplete penetrance in an LQT2 family with the A561V mutation. Only the proband (upper trace) shows QTc prolongation and prototypical morphologic changes of repolarization (T wave). Three additional members of the kindred have completely normal electrocardiograms.
Several studies have characterized the genetic epidemiology of LQTS in large cohorts of patients, and 12 different genes have been associated with the disease (Table 36–1). Notwithstanding the remarkable genetic heterogeneity, different investigators consistently showed that three genetic variants—KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3)—dominate by constituting approximately 90% of patients in whom a mutation is eventually identified (see also: www.fsm.it/cardmoc).2-4 These genes encode for the α subunits (ie, the main pore-forming proteins) of the IKs and IKr potassium channels and the cardiac sodium channel conducting INa current, respectively. Thus, the remaining genes account for a minority of patients. The immediate consequence is the availability of well-characterized genotype-based management for the most frequent forms, whereas the use of genetic information for the management of the minor LQTS variants is not as well defined due to the lack of large study cohorts.
| Variant | Gene | Protein | Effect of Mutations |
|---|---|---|---|
| LQT1 | KCNQ1 | KvLQT1 (potassium channel α subunit) | Reduced IKs |
| LQT2 | KCNH2 | HERG (potassium channel α subunit) | Reduced IKr |
| LQT3 | SCN5A | Nav1.5 (sodium channel α subunit) | Increased INa |
| LQT4 | ANK2 | Ankyrin B, anchoring protein | Reduced membrane expression of Na+ and Ca2+ channels |
| LQT5 | KCNE1 | MinK (potassium channel β subunit) | Reduced IKs |
| LQT6 | KCNE2 | MiRP (potassium channel β subunit) | Reduced IKr |
| LQT7, Andersen syndrome | KCNJ2 | Kir2.1 (potassium channel α subunit) | Reduced outward IK1 |
| LQT8, Timothy syndrome | CACNA1c | Cav1.2 (L-type calcium channel α subunit) | Increased ICα |
| LQT9 | CAV3 | Cardiac caveolin gene | Increased INa due to altered gating kinetic |
| LQT10 | SCN4B | Sodium channel β4 subunit | Reduced subunit expression causing increased INa |
| LQT11 | AKAP9 | Yotiao (KvLQT1 regulating proteins | Impaired IKs activation by catecholamines |
| LQT12 | SNTA1 | Syntrophin | Reduced NaV1.5 nitrosylation and increased current |
The final common consequence of LQTS gene mutations is the disruption of one or more ion currents that generate the cardiac action potential. Mutations may directly affect an ion channel or may hit one of their regulatory proteins. For example, an excess of sodium flowing into the cell (gain of function of INa) causes one LQTS variant (LQT3). This effect can be achieved by mutations directly affecting the sodium channel (SCN5A) gene, the sodium channel β4 subunit (SCN4B) gene, or the caveolin 3 (CAV3) gene that control the localization of the sodium channel in the plasmalemma (see Fig. 36–1 and Table 36–1). In the following sections, we address the pathophysiology of LQTS using the affected ionic current as a starting point (see Table 36–1 for a summary of LQTS genes).
Mutations in potassium channels are a frequent cause of LQTS. LQT1 is caused by mutations in the KCNQ1 gene that encode for the α subunit of the IKs current (slow component of the so-called delayed rectifier current); it accounts for 45% to 55% of patients.2,5 This current plays an important role during phase 3 of the action potential, and it is specifically sensitive to catecholamine activation. Thus, mutations in this channel become more evident during increased sympathetic tone (eg, exercise, acute emotions), with a failure to shorten action potential (ie, QT interval). This mechanism also explains the increased risk of cardiac events during exercise for LQT1 patients.6
The second most common variant of LQTS is LQT2, due to loss of function mutations in the KCNH2 gene. KCNH2 encodes for the α subunit of the IKr (rapid component of the delayed rectifier) channel, which participates in the control of cardiac repolarization, mainly during phase 3.
Overall, LQT1 and LQT2 represent the majority of LQTS patients (~60%) with an identified mutation and have a peculiar clinical presentation that should be considered for the clinical management (see Genotype-Phenotype Correlation and Clinical Management).
Additional potassium-related LQTS variants are those due to mutations in two β subunits, KCNE1-LQT5 (which coassembles with KCNQ1) and KCNE2-LQT6 (which is thought to coassemble mainly with KCNH2, but also with other potassium channels). These two genes cause LQT5 and LQT6, which together account for approximately 5% to 6% of cases. LQT5 and LQT6 have similar pathogenesis (and clinical presentation) as LQT1 and LQT2 but with a generally milder phenotype and lower penetrance.2,7 A mutation in a KCNQ1 interacting protein, AKAP9-LQT11 (also called Yotiao), has been reported in a single family.7 This mutation disrupts the cyclic adenosine monophosphate (cAMP)-mediated phosphorylation and catecholamine-dependent channel activation; thus, it is pathophysiologically similar to LQT1.
KCNJ2 causes the LQT7 variant, also known as Andersen-Tawil syndrome. The gene encodes for the cardiac inward rectifier IK1 current gene (Kir2.1) that participates in the control of the late repolarization phase and resting membrane potential. LQT7 is a rare variant (<1%) that can include, but not always, extracardiac manifestations (periodic paralysis and dysmorphic features).
SCN5A mutations causing LQT3 are due to gain of function mutations (increased late sodium current, INa) and represent the third most frequent LQTS variant. LQT3 patients have an increased risk of events at rest or during sleep. LQT3-like phenotype can also be achieved by other protein/gene mutations in rare instances. CAV3 (LQT9) mutations have been associated with this effect in 0.4% of patients.8 Anecdotal cases with a mutation in SCN4B (LQT10) and in the syntrophin gene (SNTA1, LQT12) have also been reported.9 Again, the mechanism is that of an increased late sodium current.
The following are other genes accounting for a minority of LQTS cases that have been reported (see Table 36–1):
- ANK2 (LQT4): Encodes for an intracellular protein called ankyrin, a chaperone molecule involved in ion channel localization and anchoring to the cellular membrane.
- CACNA1c (Timothy syndrome or LQT8): Encodes for the voltage-dependent L-type calcium channel (CaV1.2). This LQTS variant presents an extremely severe and rare phenotype, including QT prolongation, atrioventricular block, syndactyly, congenital heart defects, autism, reduced immune response, and developmental disorders.10 Few Timothy patients have survived after puberty in the reported series.11
The diagnosis of LQTS is based on the evaluation of the electrocardiogram (ECG) and on the measurement of the QT interval. QTc values exceeding 440 ms (in males) and 460 ms (in females) are considered abnormal. It is also evident that a “gray” area exists where QT values of normal and late QT subjects overlap (see Fig. 36–2). In these cases, diagnosis is not always straightforward, and correct use of genetic testing may be useful. Although QTc assessment is the most important step for diagnosis of LQTS, ST-T wave abnormalities (notches on the descending limb, biphasic T waves) are often evident and can support the clinical diagnosis.
Syncope is often triggered by the onset of rapid polymorphic ventricular tachycardia (VT) (torsades de pointes) that can degenerate into ventricular fibrillation and cause sudden death. The mean age of onset of symptoms is 12 years, and earlier onset is usually associated with a more severe form of the disease.
In the absence of genetic information, the single most important predictor of cardiac events is the duration of QT interval. This feature has been highlighted since the initial description of the disease,12 and it has been reproducibly confirmed thereafter in several cohorts.3,13 The evidence of QTc interval >500 ms is associated with a five-fold increased risk of events.3 Additional risk factors are female sex (approximately two-fold increased risk) and the occurrence of a first cardiac event in early childhood.14,15 In LQTS, cardiac arrhythmias are often precipitated by physical or emotional stress, although in 10% to 15% of patients, cardiac events occur at rest. Both the risk and the triggers for events are modulated by the genotype (see below). Additional increased risk is seen in the 9-month postpartum period.16
Since the mid-1970s, the evidence for an adrenergic trigger of events suggested the use of β-blockers17 to prevent cardiac arrhythmias, and the effectiveness of this approach has been constantly confirmed thereafter. Therefore, according to current guidelines,18 all LQTS patients with a history of syncope and asymptomatic individuals with definite QT prolongation should be treated with β-blockers (class I recommendation) (Table 36–2). The most frequently used drugs are nadolol (1-2.5 mg/kg/d), propranolol (2-4 mg/kg/d), and metoprolol (2-4 mg/kg/d). There are no comparative studies of efficacy on different β-blockers. Dose titration is usually based on exercise stress test, with a target of approximately 25% to 35% reduction of maximal heart rate attained on therapy. Holter monitoring is also useful to monitor for excessive nocturnal bradycardia. Lifestyle changes aimed at avoiding conditions that can precipitate arrhythmias are also indicated. Specifically, LQTS patients should avoid competitive sports, QT-prolonging drugs, and lowered potassium plasma levels (for example, during diarrhea and vomiting). An updated list of QT-prolonging drugs may be found at the Web site for the Arizona Center for Education and Research on Therapeutics (http://www.azcert.org/).
| Condition | Category | Therapy |
|---|---|---|
| LQTS | Class I | Lifestyle modification is recommended for patients with an LQTS diagnosis (clinical and/or molecular). (Level of Evidence: B) |
| β-Blockers are recommended for patients with an LQTS clinical diagnosis (ie, in the presence of prolonged QT interval). (Level of Evidence: B) | ||
| Implantation of an ICD along with use of β-blockers is recommended for LQTS patients with previous cardiac arrest and who have reasonable expectation of survival with a good functional status for more than 1 year. | ||
| In pregnant women with the LQTS who have had symptoms, it is reasonable to continue β-blocker medications throughout pregnancy and afterward, unless there are definite contraindications. (Level of Evidence: C) | ||
| Permanent pacing is indicated for sustained pause-dependent VT, with or without QT prolongation.3 | ||
| Class IIa | β-Blockers can be effective to reduce SCD in patients with a molecular LQTS analysis and normal QT interval. (Level of Evidence: B) | |
| Implantation of an ICD with continued use of β-blockers can be effective to reduce SCD in LQTS patients experiencing syncope and/or VT while receiving β-blockers and who have reasonable expectation of survival with a good functional status for more than 1 year. (Level of Evidence: B) | ||
| Class IIb | LCSD can be considered for LQTS patients with syncope, torsades de pointes, or cardiac arrest while receiving β-blockers. (Level of Evidence: B) | |
| Implantation of an ICD with use of β-blockers may be considered for prophylaxis of SCD for patients in categories possibly associated with higher risk of cardiac arrest (such as LQT2 and LQT3, QTc >500 ms) and who have reasonable expectation of survival with a good functional status for more than 1 year. (Level of Evidence: B) | ||
| CPVT | Class I | β-Blockers are indicated for patients who are clinically diagnosed with CPVT based on the presence of spontaneous or documented stress-induced ventricular arrhythmias. (Level of Evidence: C) |
| Implantation of an ICD with use of β-blockers is indicated for patients with CPVT who are survivors of cardiac arrest and who have reasonable expectation of survival with a good functional status for more than 1 year. (Level of Evidence: C) | ||
| Class IIa | β-Blockers can be effective in patients without clinical manifestations when the diagnosis of CPVT is established during childhood based on genetic analysis. (Level of Evidence: C) | |
| Implantation of an ICD with use of β-blockers can be effective for affected patients with CPVT with syncope and/or documented sustained VT while receiving β-blockers and who have reasonable expectation of survival with a good functional status for more than 1 year. (Level of Evidence: C) | ||
| Class IIb | β-Blockers can be considered for patients with CPVT who were genetically diagnosed in adulthood and never manifested clinical symptoms of tachyarrhythmias. (Level of Evidence: C) | |
| Brugada syndrome | Class I | An ICD is indicated for BrS patients with previous cardiac arrest receiving chronic optimal medical therapy and who have reasonable expectation of survival with a good functional status for more than 1 year. (Level of Evidence: C) |
| Class IIa | An ICD is reasonable for BrS patients with spontaneous ST-segment elevation in V1, V2, or V3 who have had syncope with or without mutations demonstrated in the SCN5A gene and who have reasonable expectation of survival with a good functional status for more than 1 year. (Level of Evidence: C) | |
| Clinical monitoring for the development of a spontaneous ST-segment elevation pattern is reasonable for the management of patients with ST-segment elevation induced only with provocative pharmacologic challenge with or without symptoms. (Level of Evidence: C) | ||
| An ICD is reasonable for BrS patients with documented VT that has not resulted in cardiac arrest and who have reasonable expectation of survival with a good functional status for more than 1 year. (Level of Evidence: C) | ||
| Isoproterenol should be used to treat an electrical storm in BrS. (Level of Evidence: C) | ||
| Class IIb | EP testing can be considered for risk stratification in asymptomatic BrS patients with spontaneous ST elevation with or without a mutation in the SCN5A gene. (Level of Evidence: C) | |
| Quinidine might be reasonable for the treatment of electrical storm in patients with BrS. (Level of Evidence: C) |
For the 20% to 30% of patients who experience recurrence of syncope despite β-blockade, the prophylactic implantation of an implantable cardioverter-defibrillator (ICD) should be considered. Obviously the ICD is indicated in all patients surviving cardiac arrest (secondary prevention). For primary prevention of cardiac arrest, ICD should be adopted in patients not adequately protected by β-blockers (recurrence of syncope while on therapy) and in patients with high-risk genotype (see the following section). In all instances, β-blockers should be continued after ICD implant to reduce the risk of cardiac arrest and shocks from the device. Finally, in patients with evidence of pause-dependent onset of torsade de pointes and polymorphic VT, the use of pacemaker may also be indicated.18,19 Gene-specific therapies have also been proposed to specifically counteract the effects of mutations. The only approach that has been used in the clinical setting is the administration of mexiletine in LQT3. Mexiletine, by blocking INa, may reduce the gain of function effects of LQT3 mutations. However, not all patients respond favorably due to the different biophysical consequences of the mutations.20,21 Therefore, before chronic therapy is started, this therapy should be first administered with caution in the hospital setting.
Left cardiac sympathetic denervation has also been suggested as a treatment to reduce cardiac events in patients with recurrence of cardiac events on β-blockers. Management strategies for LQTS are shown in Table 36–2.18
Several studies have contributed to shed light on genotype-phenotype correlations in LQTS. Furthermore, the progressive widespread availability of genetic testing is facilitating the implementation of genotype-phenotype correlation in clinical practice.
Gene-specific repolarization morphology and triggers for cardiac events have been reported.6,22 For example, LQT1 patients tend to experience cardiac events with physical activity, especially swimming, as opposed to LQT3 patients, who experience the majority of cardiac events at rest. Auditory stimuli and arousal are specific triggers for LQT2.23,24 Most importantly, genotype plays a relevant role in determining the risk of events and the response to β-blocker therapy. Data from international LQTS registries and referral centers have consistently shown that LQT2 and LQT3 patients have worse long-term prognosis than LQT1 patients (two-fold risk increase for LQT2 and LQT3 vs LQT1). Therefore, genotype has entered the risk stratification scheme as an independent predictor of events, together with the traditional risk factors of QT duration and sex (Fig. 36–3).3 LQTS genotype not only influences prognosis but also response to therapy. Although β-blockers are highly effective in LQT1,14,25 significantly higher recurrences of life-threatening arrhythmias are observed among LQT2 and LQT3 (hazard ratios for recurrent event in LQT2 and LQT3 vs LQT1 of 2.8 and 4.0, respectively). Because the majority of events among LQT3 patients occur at rest or during sleep, some authors have suggested that β-blockers are ineffective or may be even detrimental. Further clinical investigation is required to shed light on this issue. Incomplete efficacy of β-blockers also suggests that ICD implant for primary prevention of cardiac arrest could be considered for LQT2 and LQT3 patients with QTc >500 ms and early onset of syncope (age <7 years).
Figure 36–3
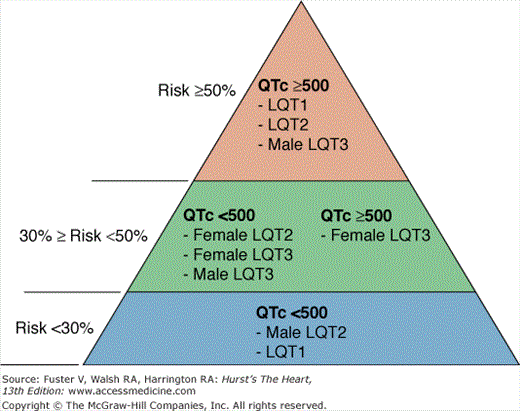
Risk stratification in long QT syndrome (LQTS). Risk categories in LQTS according to QTc duration, genotype, and sex. Percentages on the left indicate the risk of a first cardiac event (syncope or cardiac arrest) in patients younger than 40 years of age in the absence of any LQTS active treatment. Priori et al.3
It is important to point out that genotype-phenotype correlation are available for the three most prevalent LQTS variants, LQT1, LQT2, and LQT3, which represent >90% of patients in whom the genetic defect is identified. The remaining variants of LQTS account for a minority of patients, and their genotype-specific phenotype cannot be finely characterized due to the limited sample size.
Catecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a disorder of intracellular calcium handling causing adrenergic-dependent arrhythmias and sudden death. CPVT patients display an unremarkable resting ECG, but they develop a typical pattern of bidirectional or polymorphic VT that can be reproducibly elicited during exercise or catecholamine infusion (Fig. 36–4).
Figure 36–4
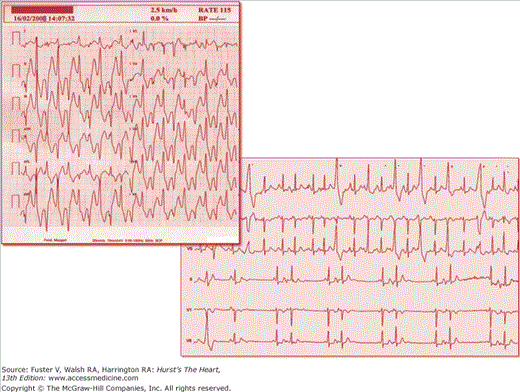
Arrhythmias in catecholaminergic polymorphic ventricular tachycardia (CPVT). The upper panel shows bidirectional ventricular tachycardia during exercise stress test at low workload and arising at a sinus heart rate of 115 beats/min. An example of supraventricular arrhythmias is shown in the lower panel.
Two genetic variants of CPVT have been identified by linkage analysis and candidate gene screening. The autosomal dominant variant (CPVT1) is largely more prevalent (~50% of patients) and is due to mutations in the RyR2 gene, encoding for the cardiac ryanodine receptor.26 The ryanodine receptor is a tetrameric intracellular Ca2+ release channel spanning the membrane of the sarcoplasmic reticulum (SR), and it is required for excitation-contraction coupling (see Fig. 36–1).
The autosomal recessive variant (CPVT2) maps to the short arm of chromosome 1 and is due to mutations of the calsequestrin 2 gene (CASQ2).27 Data from our CPVT registry suggest that CASQ2 mutations account for 3% to 5% of all genotyped patients.28 Calsequestrin is a calcium-buffering protein primarily expressed in the terminal cisternae of the SR. It binds Ca2+ ions to control the free Ca2+ concentration in the SR, and it also directly modulates the RyR2 open probability (presumably through physical interaction with RyR2 channels mediated by triadin and junctin that constitute parts of the SR calcium release macromolecular complex).
More recently, it has been proposed that mutations of two other genes may cause an arrhythmogenic disorder in the structurally normal heart that resembles the classical description of CPVT (phenocopies).
KCNJ2, which is more frequently associated with Andersen syndrome (LQT7), may cause bidirectional VT with mild or absent QT prolongation and no extracardiac involvement. We have shown in vitro29 that the cellular mechanism of KCNJ2 CPVT is similar to that of the typical RyR2 CPVT, with the appearance of delayed afterdepolarizations (DADs) during β-adrenergic stimulation (see following section). The differential diagnosis between KCNJ2 CPVT and RyR2 CPVT is relevant because KCNJ2 mutations are usually milder and sudden death is an exceptional event.30KCNJ2 screening may be considered only in RyR2– CASQ2–negative CPVT cases.
Anecdotal reports show that ankyrin B mutations may cause a CPVT phenotype31; although there is no systematic assessment of the relative prevalence of this variant, it is probably rare, and there is current no indication for a systematic screening of this gene in CPVT patients.



