Chapter 54 Genetics and Cardiac Arrhythmia Syndromes
Cardiac arrhythmias are major causes of morbidity and mortality, including sudden cardiac death (SCD). SCD in the United States occurs with a reported incidence of more than 300,000 persons per year.1 Although coronary heart disease is a major cause of death, other etiologies contribute to this problem. In many of these non–ischemia-related cases, autopsies are unrevealing. Interest in identifying the underlying cause of the death in these instances has been focused on cases of unexpected arrhythmogenic death, which is estimated to represent 5% of all SCDs. In cases in which no structural heart disease can be identified, long QT syndrome (LQTS), ventricular pre-excitation (Wolff-Parkinson-White syndrome), and idiopathic ventricular fibrillation (IVF) or Brugada syndrome (characterized by ST-segment elevation in the right precordial leads with or without right bundle branch block [RBBB]) are most commonly considered as likely causes.1–3 Another important disease in which arrhythmias are believed to play a central role is sudden infant death syndrome (SIDS), a disorder with no structural abnormalities.4
Arrhythmogenic right ventricular dysplasia (ARVD) is also a significant cause of SCD and is considered a primary electrical disease despite being associated with fibrosis and fatty infiltration of the right ventricle.5 The arrhythmias associated with ARVD also occur in other disorders in which structurally normal myocardium is seen, such as catecholaminergic ventricular tachycardia (VT).6
This chapter describes the current understanding of the clinical and molecular genetic aspects of inherited diseases in which arrhythmias are prominent features. The discussion should serve as an introduction and overview to these conditions. Newer disorders are rapidly being added to this list and are discussed in more detail elsewhere in this textbook. A more detailed treatment of the current state of knowledge regarding the molecular basis and basic electrophysiological mechanisms of inherited arrhythmia syndromes is discussed in Chapters 6 and 7. Individual clinical syndromes are discussed in Chapters 62 to 65.
Long QT Syndrome
Clinical Description
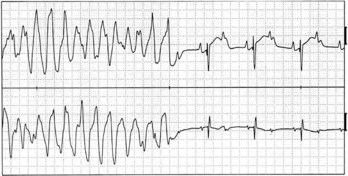
LQTS is an inherited or acquired disorder of repolarization identified by the electrocardiographic abnormalities of prolongation of the Q-T interval corrected for heart rate (QTc), usually above 460 to 480 ms; relative bradycardia; T-wave abnormalities (Figure 54-1); and episodic ventricular tachyarrhythmias, particularly torsades de pointes (Figure 54-2).7 The inherited form of LQTS is transmitted as an autosomal dominant or autosomal recessive trait. Acquired LQTS may be seen as a complication of various drug therapies or electrolyte abnormalities. Whether the abnormality is genetic or acquired, the clinical presentation is similar.1,7 The initial presentation of LQTS is heterogeneous and most commonly includes syncope, which, in many instances, is triggered by emotional stress, exercise, or auditory phenomena. Other presenting features include seizures or palpitations. SCD is the first symptom in some individuals, but some other cases are diagnosed by surface electrocardiogram (ECG) as a family screening evaluation necessitated by family history of LQTS or SCD.

Clinical Genetics
Two differently inherited forms of familial LQTS have been reported. Romano-Ward syndrome is the most common of the inherited forms of LQTS and appears to be transmitted as an autosomal dominant trait.8,9 In this disorder, the disease gene is transmitted to 50% of the offspring of an affected individual. However, low penetrance has been described, so gene carriers may, in fact, have no clinical features of disease.10 Individuals with Romano-Ward syndrome have the pure syndrome of prolonged Q-T interval on ECG, with the associated symptom complex of syncope, SCD, and, in some patients, seizures.11,12 Occasionally, other noncardiac abnormalities such as diabetes mellitus, asthma, or syndactyly may also be associated with QT prolongation.13–16 LQTS may also be involved in some cases of SIDS, which, in some cases, appear in several family members.5,17,18
Jervell and Lange-Nielsen syndrome (JLNS) is a relatively uncommon inherited form of LQTS. Classically, this disease has been described as having apparent autosomal recessive transmission.19–21 These patients have a clinical presentation identical to that in patients with Romano-Ward syndrome but also have associated sensorineural deafness. Clinically, patients with JLNS usually have longer Q-T intervals compared with individuals with Romano-Ward syndrome and also have a more malignant course. Priori and colleagues have reported autosomal recessive cases of Romano-Ward syndrome as well, thus changing one of the sine qua non of JLNS.22
Gene Identification in Romano-Ward Syndrome
KVLQT1 or KCNQ1: The LQT1 Gene
The first of the genes mapped for LQTS, termed LQT1, required 5 years from the time that mapping to chromosome 11p15.5 was first reported to gene cloning.23 This gene, originally named KVLQT1, but more recently called KCNQ1 (Table 54-1), is a novel potassium channel gene that consists of 16 exons, spans approximately 400 kb, and is widely expressed in human tissues, including the heart, inner ear, kidney, lung, placenta, and pancreas but not in the skeletal muscle, liver, or brain.24 Although most of the mutations are “private” (i.e., only seen in one family), at least one frequently mutated region (called a “hot spot”) of KVLQT1 exists.25–27 This gene is the most commonly mutated gene in LQTS.
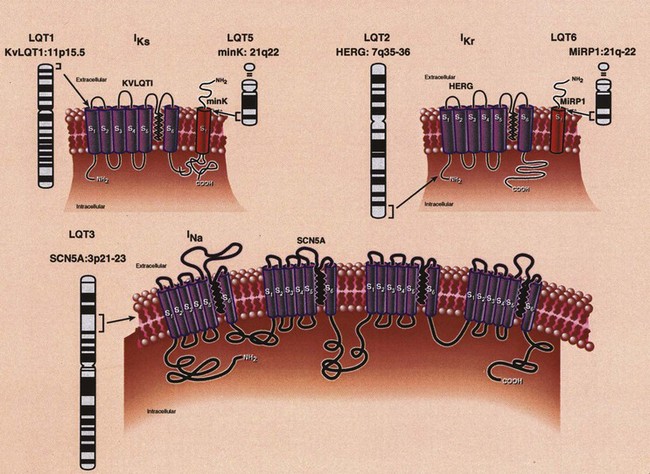
Analysis of the predicted amino acid sequence of KVLQT1 suggests that it encodes a potassium channel α-subunit with a conserved potassium-selective pore-signature sequence flanked by six membrane-spanning segments similar to shaker-type channels (Figure 54-3).24,27–29 A putative voltage sensor is found in the fourth membrane-spanning domain (S4), and the selective pore loop is located between the fifth and sixth membrane-spanning domains (S5,S6). Biophysical characterization of the KVLQT1 protein confirmed that KVLQT1 is a voltage-gated potassium channel protein subunit, which requires coassembly with a β-subunit called minK to function properly.28,29 Expression of either KVLQT1 or minK alone results in either inefficient or no current development. When minK and KVLQT1 are co-expressed in either mammalian cell lines or Xenopus oocytes, however, the slowly activating potassium current (IKs) is developed in cardiac myocytes.28,29 The combination of normal and mutant KVLQT1 subunits forms abnormal IKs channels, and these mutations are believed to act through a dominant-negative mechanism (the mutant form of KVLQT1 interferes with the function of the normal wild-type form through a “poison pill” type mechanism) or a loss-of-function mechanism (only the mutant form loses activity).30
Because KVLQT1 and minK form a unit, mutations in minK could also be expected to cause LQTS. This fact was subsequently demonstrated (discussed below).31
HERG or KCNH2: The LQT2 Gene
The LQT2 gene was initially mapped to chromosome 7q35-36 by Jiang et al, and subsequently, candidate gene screening identified the disease-causing gene HERG (human ether-a-go-go-related gene), a cardiac potassium channel gene to be the LQT2 gene (see Table 54-1).27,32 HERG was originally cloned from a brain cDNA library and found to be expressed in neural crest–derived neurons, microglia, a wide variety of tumor cell lines, and the heart.33–37 LQTS-associated mutations were identified in HERG throughout the gene, including missense mutations, intragenic deletions, stop codons, and splicing mutations.27,37,38 Currently, this gene is thought to be the second most common gene mutated in LQTS (second to KVLQT1). As with KVLQT1, “private” mutations that are scattered throughout the entire gene without clustering preferentially are seen.
HERG consists of 16 exons and spans 55 kb of genomic sequence.37 The predicted topology of HERG (see Figure 54-3) is similar to that of KVLQT1. Unlike KVLQT1, HERG has extensive intracellular amino-and-carboxyl termini, with a region in the carboxyl-terminal domain having sequence similarity to nucleotide binding domains (NBDs).
Electrophysiological and biophysical characterization of expressed HERG in Xenopus oocytes established that HERG encodes the rapidly activating delayed rectifier potassium current IKr.39–41 Electrophysiological studies of LQTS-associated mutations showed that they act through either a loss of function or a dominant negative mechanism.41,42 In addition, protein trafficking abnormalities have been shown to occur.43,44 This channel has been shown to coassemble with β-subunits for normal function, similar to that seen in IKs. McDonald et al initially suggested that the complexing of HERG with minK is needed to regulate the IKr potassium current.45 Bianchi et al provided confirmatory evidence that minK is involved in the regulation of both IKs and IKr.46 Abbott et al identified MiRP1 as a β-subunit for HERG (discussed below).47
SCN5A: The LQT3 Gene
The positional candidate gene approach was also used to establish that the gene responsible for chromosome 3–linked LQTS (LQT3) is the cardiac sodium channel gene SCN5A (see Table 54-1).48,49 SCN5A is highly expressed in the human myocardium and brain but not in the skeletal muscle, liver, or uterus.50–52 It consists of 28 exons that span 80 kb and encodes a protein of 2016 amino acids with a putative structure that consists of four homologous domains (DI to DIV), each of which contains six membrane-spanning segments (S1 to S6) similar to the structure of the potassium channel α-subunits (see Figure 54-3).27,39 Linkage studies with LQT3 families and SCN5A initially demonstrated linkage to the LQT3 locus on chromosome 3p21-24, and multiple mutations were subsequently identified.50,51 Biophysical analysis of the initial three mutations were expressed in Xenopus oocytes, and it was found that all mutations generated a late phase of inactivation-resistant, mexiletine- and tetrodotoxin-sensitive whole-cell currents through multiple mechanisms.53,54 Two of the three mutations showed dispersed reopening after the initial transient current, but the other mutation showed both dispersed reopening and long-lasting bursts.54 These results suggested that SCN5A mutations act through a gain of function mechanism (the mutant channel functions normally, but with altered properties such as delayed inactivation) and that the mechanism of chromosome 3–linked LQTS is persistent non-inactivated sodium current in the plateau phase of the action potential. Later, An et al showed that not all mutations in SCN5A are associated with persistent current and demonstrated that SCN5A interacted with β-subunits.55
minK or KCNE1: The LQT5 Gene
minK (IsK or KCNE1) was initially localized to chromosome 21 (21q22.1) and found to consist of three exons that span approximately 40 kb (see Table 54-1). It encodes a short protein consisting of 130 amino acids and has only one transmembrane-spanning segment with small extracellular and intercellular regions (see Figure 54-3).30,31,56 When expressed in Xenopus oocytes, it produces potassium current that closely resembles the slowly activating delayed-rectifier potassium current IKs in cardiac cells.56,57 The fact that the minK clone was only expressed in Xenopus oocytes and not in mammalian cell lines raised the question whether minK is a human channel protein. With the cloning of KVLQT1 and the coexpression of KVLQT1 and minK in both mammalian cell lines and Xenopus oocytes, it became clear that KVLQT1 interacts with minK to form the cardiac slowly activating delayed rectifier IKs current.28,29 minK alone cannot form a functional channel but induces the IKs current by interacting with endogenous KVLQT1 protein in Xenopus oocytes and mammalian cells. Bianchi et al showed that mutant minK results in abnormalities of IKs, IKr as well as protein trafficking abnormalities.46 McDonald et al showed that minK also complexes with HERG to regulate the IKr potassium current.45 Splawski et al demonstrated that minK mutations cause LQT5 when they identified mutations in two families with LQTS.31 In both cases, missense mutations (S74L, D76N) were identified; they reduced IKs by shifting the voltage dependence of activation and accelerating channel deactivation. This was supported by the fact that a murine model of minK-defective LQTS was also created.58 The functional consequences of these mutations include delayed cardiac repolarization and, hence, an increased risk of arrhythmias.
MiRP1 or KCNE2: The LQT6 Gene
MiRP1, the minK-related peptide 1, or KCNE2 (see Table 54-1), is a novel potassium channel gene recently cloned and characterized by Abbott and colleagues.47 This small integral membrane subunit protein assembles with HERG (LQT2) to alter its function, enabling full development of the IKr current (see Figure 54-3). MiRP1 is a 123–amino acid channel protein with a single predicted transmembrane segment similar to that described for minK.56 Chromosomal localization studies mapped this KCNE2 gene to chromosome 21q22.1, within 79kb of KCNE1 (minK) and arrayed in opposite orientation.47 The open reading frames of these two genes share 34% identity, and both are contained in a single exon, suggesting that they are related through gene duplication and divergent evolution.
Three missense mutations associated with LQTS and ventricular fibrillation (VF) were identified in KCNE2 by Abbott et al, and biophysical analysis demonstrated that these mutants form channels that open slowly and close rapidly, thus diminishing potassium currents.47 In one case, the missense mutation, a C-to-G transversion at nucleotide 25, which produced a glutamine (Q) to glutamic acid (E) substitution at codon 9 (Q9E) in the putative extracellular domain of MiRP1, led to the development of torsades de pointes and VF after intravenous clarithromycin infusion (i.e., drug induced).
Genetics and Physiology of Autosomal Recessive Long QT Syndrome (Jervell and Lange-Nielsen Syndrome)
Neyroud et al reported the first molecular abnormality in patients with JLNS when they reported on two families in which three children were affected by JLNS and in whom a novel homozygous deletion-insertion mutation of KVLQT1 was found.59 A deletion of 7 bp and an insertion of 8 bp at the same location led to premature termination at the C-terminal end of the KVLQT1 channel. At the same time, Splawski et al identified a homozygous insertion of a single nucleotide that caused a frameshift in the coding sequence after the second putative transmembrane domain (S2) of KVLQT1.60 Together, these data strongly suggested that at least one form of JLNS is caused by homozygous mutations in KVLQT1 (see Table 54-1). This has been confirmed by others.27,30,61,62
As a general rule, heterozygous mutations in KVLQT1 cause Romano-Ward syndrome (LQTS only), whereas homozygous (or compound heterozygous) mutations in KVLQT1 cause JLNS (LQTS and deafness). The hypothetical explanation suggests that although heterozygous KVLQT1 mutations act by a dominant-negative mechanism, some functional KVLQT1 potassium channels still exist in the stria vascularis of the inner ear. Therefore congenital deafness is averted in patients with heterozygous KVLQT1 mutations. For patients with homozygous KVLQT1 mutations, no functional KVLQT1 potassium channels can be formed. It has been shown by in situ hybridization that KVLQT1 is expressed in the inner ear, suggesting that homozygous KVLQT1 mutations can cause the dysfunction of potassium secretion in the inner ear and lead to deafness.60 However, it should be noted that incomplete penetrance exists, and not all heterozygous or homozygous mutations follow this rule.11,22
As with Romano-Ward syndrome, if KVLQT1 mutations can cause the phenotype, it could be expected that minK mutations could also be causative of the phenotype (JLNS). Schulze-Bahr et al, in fact, showed that mutations in minK result in JLNS syndrome as well, and this was confirmed subsequently (see Table 54-1).60,63 Hence, abnormal IKs current, whether caused by homozygous or compound heterozygous mutations in KVLQT1 or mink, results in LQTS and deafness.
Genotype-Phenotype Correlations in Long QT Syndrome
Clinical Features
Kimbrough et al recently reported on the study of 211 probands with LQTS and classified the severity in the probands, affected parents, and siblings.64 Importantly, they showed that the severity of the disease in the proband did not correlate with the clinical severity seen in first-degree relatives, specifically their parents and siblings. In fact, variable intrafamily penetrance was noted, consistent with other genetic and environmental factors playing a role in modulating and modifying clinical manifestations in members of the same family. Several stratifiers were identified as important.
These findings complement the findings previously described by Zareba et al.65 In this study, the authors provided evidence of clinical outcome, age of onset, and frequency of events based on genotype. Patients with mutations in LQT1 had the earliest onset of events and the highest frequency of events followed by mutations in LQT2. The risk of SCD in these two groups was relatively low for any event. Mutations in LQT3 resulted in a paucity of syncopal events, but events commonly resulted in SCD. In addition, mutations in LQT3 resulted in the longest QTc duration. Mutations in LQT1 and LQT2 appeared to be associated with stress-induced symptoms, with LQT1 associated with exercise and swimming and LQT2 associated with auditory triggers.66–71 LQT3 appeared to be associated with sleep-associated symptoms and events.
Electrocardiographic and Biophysical Features
In 1995, Moss and colleagues provided the first evidence that mutations in different genes cause differing ECG features.72 Specifically, these authors focused on the different types of T waves seen in patients with LQT1 versus LQT2 versus LQT3. ECGs of patients with LQT1 were shown to display broad-based T waves, those with LQT2 had low-amplitude T waves, and those with LQT3 mutations had distinctive T waves with late onset. More recently, Zhang et al showed that there are actually four different ST-T wave patterns.73 Using these definitions, they were able to identify 88% of patients with LQT1 and LQT2 accurately by surface ECG and 65% of LQT3 carriers. Prospectively, these authors correctly predicted the genotype in 100% of patients.
Further insight into ECG findings and genotype were reported by Lupoglazoff et al using Holter monitor analysis.74 Analysis of 133 patients with LQT1 57 LQT2 carriers, and 100 control individuals, led the authors to conclude that T wave morphology was normal in most patients with LQT1 (92%) and in normal controls (96%), but the vast majority of patients with LQT2 had abnormal T waves (19% normal, 81% abnormal). In the largest percentage of patients with LQT2, T-wave notching was identified, with the T-wave protuberance seen above the horizontal, whereas another subset had a bulge at or below the horizontal. In the former case, young age, missense LQT2 mutations, and mutations in the core domain of HERG predicted morphology, whereas potential diagnostic clues gained by the latter morphology included amino-terminal or carboxy-terminal mutations or frameshifts in HERG.
Animal Models of Long QT Syndrome
By using an arterially perfused canine left ventricular wedge preparation developed pharmacologically, induced animal models of LQT1, LQT2, and LQT3 have been created.75,76 By using chromanol 293B, a specific IKs blocker, a model that mimics LQT1 was produced.75 In this model, IKs deficiency alone was not enough to induce torsades de pointes, but the addition of β-adrenergic influence (i.e., isoproterenol) predisposed the myocardium to torsade by increasing trans-mural dispersion of repolarization. The addition of a β-blocker or mexiletine reduced the ability to induce torsades de pointes, suggesting that these medications might improve patient outcomes.
Models for LQT2 and LQT3 were created by using D-sotalol (LQT2) or ATX-II (LQT3) in this wedge preparation.76 Both drugs preferentially prolong M cell action potential duration (APD), with ATX-II also causing a sharp rise in trans-mural dispersion. Mexiletine therapy abbreviated the Q-T interval prolongation in both models and reduced dispersion. Spontaneous torsades de pointes was suppressed, and the vulnerable window during which torsades de pointes induction occurs was also reduced in both models. These models support the current understanding of the different subtypes of LQTS and provide an explanation for potential therapies.
Therapeutic Options in Long QT Syndrome
Currently, the standard therapeutic approach in LQTS is the initiation of β-blockers at the time of diagnosis.7 Recently, Moss et al demonstrated significant reduction in cardiac events using β-blockers.77 However, syncope, aborted cardiac arrest, and SCD do continue to occur. When β-blockers cannot be used, such as in patients with asthma, other medications such as mexiletine have been tried.78 When medical therapy has failed, left sympathectomy or therapy with an implantable cardioverter-defibrillator (ICD) has been used.7
Genetics-based therapy has also been described. Schwartz et al showed that sodium channel blocking agents (i.e., mexiletine) shorten the QTc in patients with LQT3, whereas exogenous potassium supplementation or potassium channel openers have been shown to be potentially useful in patients with potassium channel defects.78–80 However, long-term potassium therapy with associated potassium-sparing agents has been unable to keep the serum potassium above 4 mmol/L because of renal potassium homeostasis. This suggests that long-term potassium therapy may not be useful. In addition, no definitive evidence that these approaches (i.e., sodium channel blockers, exogenous potassium, or potassium channel openers) improve survival has been published.
Andersen Syndrome (LQT7)
Clinical Aspects
Andersen and colleagues (1971)81 identified a complex phenotype, including ventricular arrhythmias, potassium-sensitive periodic paralysis, and dysmorphic features. The dysmorphisms included hypertelorism, broad nasal root, defects of the soft and hard palate, as well as short stature. More recently, skeletal abnormalities have broadened the phenotype (Andelfinger et al).82 The associated cardiac abnormalities include QTC prolongation, ventricular tachycardia (VT), ventricular fibrillation (VF), and atrial arrhythmias. Torsades de pointes and bi-directional VT have been seen. In addition, repolarization abnormalities affecting late repolarization and resembling giant U waves are common. SCD has not been reported as a major risk in this disorder.
Genetic Aspects
Andersen syndrome was originally mapped to chromosome 17q23-q24.2 by Plaster et al83 who used genome-wide linkage analysis. The critical region within this locus was narrowed, and candidate gene mutation screening identified mutations in KCNJ2, which encodes an inward rectifier potassium channel called Kir2.1 (Tristani-Firouzi et al).84 This channel is highly expressed in the heart and plays a role in phase 3 repolarization and in the resting membrane potential. Multiple gene mutations have been identified, to date, with relatively high penetrance noted. Functional studies have demonstrated reduction or suppression of Ik1, by a haplo insufficiency or dominant negative effect. This gene may play a role in developmental signaling pathways as well, which is believed to cause dysmorphisms.
Brugada Syndrome
Clinical Aspects of Brugada Syndrome
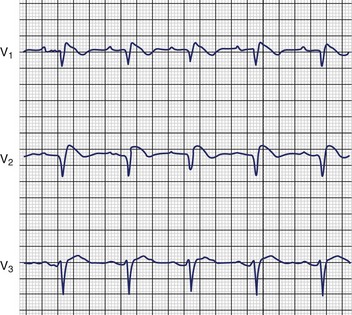
The first identification of the electrocardiographic pattern of RBBB with ST elevation in leads V1 to V3 was reported by Osher and Wolff.85 Shortly thereafter, Edeiken identified persistent ST elevation without RBBB in 10 asymptomatic males, and Levine et al described ST elevation in the right chest leads and conduction block in the right ventricle in patients with severe hyperkalemia.86,87 The first association of this ECG pattern with SCD was described by Martini et al and later by Aihara et al.88,89 This association was further confirmed in 1991 by Pedro and Josep Brugada, who described four patients with SCD and aborted SCD, with ECGs demonstrating RBBB and persistent ST elevation in leads V1 to V3 (Figure 54-4).90 In 1992 these authors characterized what they believed to be a distinct clinical and electrocardiographic syndrome.3
The finding of ST elevation in the right chest leads has been observed in various clinical and experimental settings and is not unique to or diagnostic of Brugada syndrome by itself.91 Situations in which these ECG findings occur include electrolyte or metabolic disorders, pulmonary or inflammatory diseases, and abnormalities of the central or peripheral nervous system. In the absence of these abnormalities, the term idiopathic ST elevation is often used and may identify patients with Brugada syndrome.
The ECG findings and associated sudden and unexpected death have been reported as common problems in Japan and Southeast Asia, where it most commonly affects men during sleep.92 This disorder, known as sudden and unexpected death syndrome (SUDS) or sudden unexpected nocturnal death syndrome (SUNDS), has many other names in Southeast Asia: bangungut (to rise and moan in sleep) in the Philippines; non-laitai (sleep-death) in Laos; lai-tai (died during sleep) in Thailand; and pokkuri (sudden and unexpectedly ceased phenomena) in Japan. General characteristics of SUNDS include young, healthy males in whom sudden death, preceded by a groan, occurs usually during sleep late at night. No precipitating factors are identified, and autopsy findings show no structural heart disease.93 Life-threatening ventricular tachyarrhythmias as a primary cause of SUNDS have been demonstrated, with VF occurring in most cases.94
The risk of SCD associated with Brugada syndrome and SUNDS in European and Southeast Asian individuals has been reported to be extremely high; approximately 75% of patients, as reported by Brugada et al, survived cardiac arrest.3,90,95 In addition, symptomatic and asymptomatic patients have been considered to be at equal risk. Priori et al have, however, disputed this claim.96 In a study of 60 patients with Brugada syndrome, asymptomatic patients had no episodes or events. The importance of this difference is its impact on therapeutic decision making, as currently all patients receive ICD therapy. Should the data of Priori et al hold up, selective use of ICDs would be appropriate.96 If selective use of ICDs were to be considered, other diagnostic tests for risk stratification would be necessary.
Kakishita et al studied a high-risk group of patients, 37% of whom had experienced spontaneous episodes of VF.97 As the majority of patients had ICDs, the authors were able to show that 65% of episodes were preceded by premature ventricular complexes (PVCs), which were essentially identical to the initiating PVCs of VF in morphology. In fact, the PVCs initiating all VF episodes arose from the terminal portions of the T wave, and pause-dependent arrhythmias were rare. This suggests that vigilant evaluation by Holter monitoring could identify at-risk patients. In addition, the authors suggested that the use of radiofrequency (RF) ablation targeting the initiating PVCs could be helpful in reducing risk and reducing the need for ICD placement.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


