Chapter 65 Genetic Diseases
Catecholaminergic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is one of the most malignant inherited arrhythmogenic diseases, with a high incidence of sudden cardiac death among affected individuals. The first report of a patient with this disease was published in 1975, but the first systematic description came in 1978 with the work of Coumel et al and was further refined by the same group in 1995.1–3 In 2001, the authors of this chapter reported for the first time that the autosomal dominant form of the disease was caused by mutations in the gene of the cardiac ryanodine receptor.4 Shortly thereafter, the gene for an autosomal recessive form of CPVT was identified as the gene encoding cardiac calsequestrin5; overall, it became clear that CPVT is caused by abnormal calcium (Ca2+) homeostasis.
Clinical Presentation and Diagnosis
The initial presentation of CPVT is syncope or cardiac arrest occurring during adrenergic stress.3,4 The mean age of onset of the first syncope is 12 years.6–8 In the absence of treatment, the disease is highly lethal: The estimated incidence of sudden death is of 30% before age 40 years.9 Sudden death may be the first manifestation of the disease, thus making prevention of lethal events a difficult task.
Minor, nondiagnostic features are resting sinus bradycardia and prominent U waves, but their diagnostic value has never been systematically addressed.3,10 Furthermore, mild QT prolongation in some CPVT cases has been reported2,3; thus the differential diagnosis for CPVT should include LQTS. Some patients with LQTS do have a mild phenotype (borderline QT interval and no symptoms), but their prognosis is completely different from that of patients with CPVT, which presents a higher incidence of sudden death and a limited response to β-blocker therapy.6,11
In the authors’ series of patients with CPVT, a positive history of sudden death, stress-induced syncope, or seizures was present in 30% of patients, suggesting that careful collection of family history plays an important role in the correct identification of this condition.6
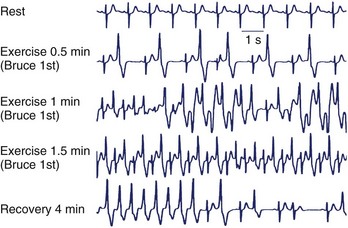
Exercise stress testing is the single most important tool, since CPVT arrhythmias are often reproducible during graded exercise (Figure 65-1). The typical behavior of CPVT arrhythmias is that of a progressive worsening on increase in workload; isolated premature beats or couplets initially appear between 90 and 110 beats per minute (beats/min) followed by runs of nonsustained or sustained ventricular tachycardia (VT) when the heart rate further increases (see Figure 65-1).12 Supraventricular arrhythmias are also a common finding and often precede the onset of ventricular arrhythmias.13 The morphology of VT is a hallmark of the disease: the so-called bidirectional VT, which is characterized by a 180-degree beat-to-beat rotation of the axis of the QRS complexes on the frontal plane.4,6 Although this pattern is recognizable in the majority of patients, it is important to be aware that some patients also present with irregular polymorphic VT. Furthermore, the initial presentation of the disease may also be that of ventricular fibrillation (VF) triggered by sudden adrenergic activation that may be interpreted as idiopathic VF in the absence of documentation of typical CPVT arrhythmias.6 Holter monitoring and implantable loop recorders are helpful for diagnosis, especially in those patients in whom emotional triggers (alone or in combination with exercise) are more arrhythmogenic than exercise alone.14,15
Genetic Bases
The majority of familial CPVT cases present an autosomal dominant pattern of inheritance. In 1999, Swan et al identified a significant linkage with some microsatellite markers at locus 1q42-q43.16 On the basis of this information, the authors of this chapter undertook a candidate gene screening in the critical region and identified the cardiac ryanodine receptor (RyR2) as the mutant CPVT gene.4 In the same year, Lahat et al described an autosomal recessive transmission pattern on the chromosomal locus 1p13-21 in a large Bedouin CPVT family.17 Subsequently, they also succeeded in identifying a mutation on the cardiac calsequestrin (CASQ2) gene, encoding for cardiac calsequestrin.5
Autosomal dominant (RyR2) CPVT is, by far, the most frequent variant, and CASQ2 mutations represent 1% to 2% of all genotyped CPVT. More recently, on the basis of the evidence that the patients with Andersen-Tawil syndrome may have bi-directional VT, it has been suggested that some CPVT cases can be explained by KCNJ2 mutations.18,19 A detailed discussion of the link between KCNJ2 mutations and CPVT is beyond the scope of this chapter, but it is relevant to note here that preliminary experimental studies on adenoviral-infected cardiac myocytes with KCNJ2-CPVT mutations show a cellular phenotype very similar to that observed in RyR2-mutant myocytes.20
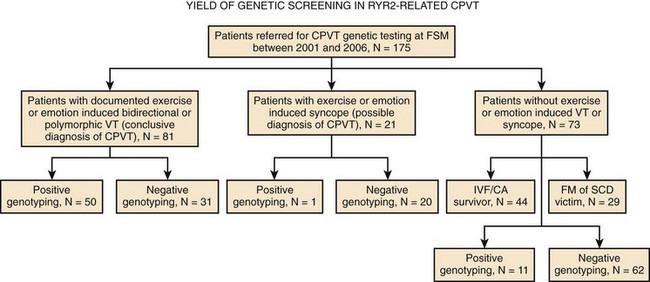
In 2007, a new autosomal recessive form of CPVT mapping on the chromosomal locus 7p14-22 was reported by Bhuiyan et al, but the gene disease has not yet been discovered.21 So far, more than 70 different mutations have been associated with CPVT, and they all are single–base pair substitutions causing the replacement of an amino acid. As expected with regard to autosomal recessive disorders, the number of families with CPVT linked to CASQ2 mutations is fairly small. At present, only seven mutations have been discovered, and they can be inherited in homozygous or compound heterozygous form. A recent analysis from the authors’ group has demonstrated that genetic screening on the RyR2 gene can identify at least 62% of probands who present with a clinical picture suggestive for the disease22; genetic testing helps identify silent mutation carriers and for reproductive counseling (Figure 65-2). Furthermore, RyR2-negative and CASQ2-negative patients should undergo KCNJ2 screening. KCNJ2 mutations are associated with a favorable outcome (sudden death is an exceptional event in this case).
Mechanisms of Arrhythmias in Autosomal Dominant CPVT: From Cellular Studies to Engineered Murine Models
Summary of RyR2 Physiology
Since Ca2+ is not only important for electrical and mechanical activation but also for a wide range of enzymatic and metabolic pathways, it is clear that RyR2 abnormalities may exert additional (and, so far, not thoroughly investigated) consequences on cell physiology.23
RyR2 Mutations and Catecholaminergic Polymorphic Ventricular Tachycardia
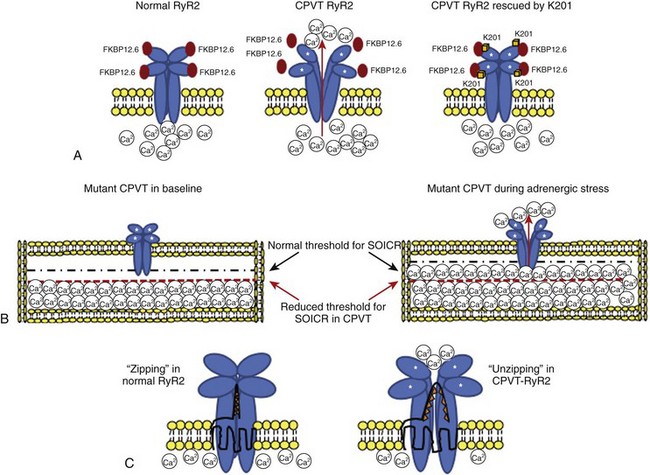
The proposed “primum movens” for RyR2-CPVT mutations to lead to Ca2+ overload is uncontrolled release (leakage) during diastole, which could be detected, according to different authors, only on adrenergic activation (phosphorylation) or already in the unstimulated condition.24–26 Given the complexity of the SR Ca2+ release process, the leakage could, in principle, be caused by several mechanisms.24,25,27 The hypotheses so far advanced are summarized in the following paragraphs (Figure 65-3).
Subcellular Mechanisms for RyR2 Dysfunction in Catecholaminergic Polymorphic Ventricular Tachycardia
FKBP12.6 Dissociation with RyR2 Mutants
FKBP12.6, also known as calstabin, is thought to play a role as a “stabilizer” of the channel in the closed state during diastole. Hints for the possible role of FKBP12.6 in CPVT were initially collected in experimental studies in heart failure. During congestive heart failure, chronic adrenergic hyperactivation may lead to weaker FKBP12.6 binding that can be the cause of Ca2+ overload-mediated arrhythmogenesis in this common disease.28 The FKBP12.6 hypothesis, originally developed by Andrew Marks and co-workers, was subsequently extended to CPVT pathophysiology; these authors demonstrated that some RyR2 mutants may cause a reduced binding affinity for FKBP12.6, which is further increased by sympathetic stimulation (see Figure 65-3).27 More recently, the same group performed an FKBP12.6 binding assay in a RyR2 knock-in mouse engineered with the R2474S mutation which has a CPVT-like phenotype29; they confirmed their initial hypothesis by demonstrating reduced FKBP12.6 binding in the presence of adrenergic stimulation. In parallel with these studies, Marks’ group also tried to pharmacologically treat the binding defect by using K201, a benzothiazepine derivative that was expected to increase FKBP-RyR2 binding. Long-term administration of this drug (or of its novel derivative, S107) was able to restore binding and to prevent the occurrence of arrhythmias both in the FKBP12.6 knock-out murine model and in the RyR2R2474S/WT knock-in murine model.29,30
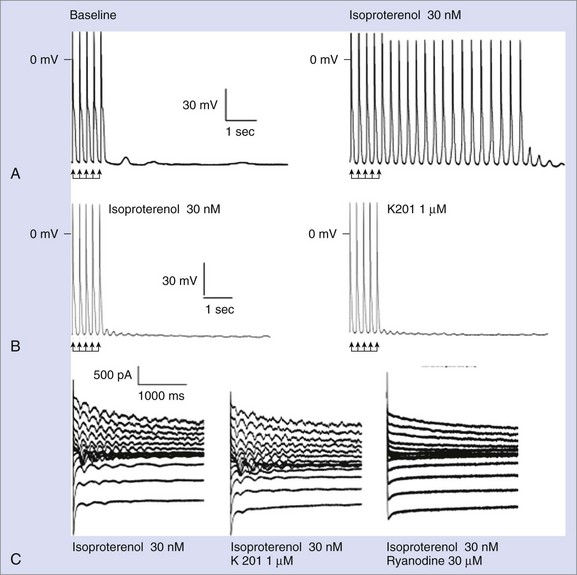
Despite the large amount of evidence provided by the Andrew Marks group, other authors either were not able to replicate their results or provided conflicting evidence when they used alternative approaches. George et al and Jiang et al reported normal FKBP12.6-RyR2 interaction in vitro.24,31 Chronic treatment with K201 did not prevent arrhythmias in vivo or the occurrence of DADs in isolated myocytes in a CPVT knock-in mouse RyR2R4496C/WT in a study by the authors of this chapter (Figure 65-4).32 Furthermore, in this model, Western blot experiments also demonstrated physiological interaction between RyR2 and FKBP12.6.32 Along these lines, Zissimopulos et al demonstrated that FKBP12.6 binding is either unaltered or even increased in the presence of CPVT-RyR2 mutations.33 Another report by Hunt et al provided evidence that K201 may modulate RyR2 independently from FKBP12.6.34 For example, these authors showed that K201 abolished spontaneous Ca2+ release in cardiac myocytes in the presence of FK506 that dissociates FKBP12.6 from RyR2.
Defective Interdomain Interaction
RyR2 is a large molecule with a complex tertiary and quaternary structure, in which interactions between domains are important for channel function. Indeed, domain-domain interactions ensure proper folding during the opening and closing of the channel.35
Several RyR2-CPVT mutations occur in the N-terminal (residue 70-466) and the central domains (residues 2246-2543).36 The N-terminal and central domains interact, and their relative conformational change from zipped (tight interaction) to unzipped (loose interaction) is involved in the channel gating process.35 Recent data have highlighted how some CPVT-related mutants may affect this mechanism by inducing a shift toward a mostly unzipped interaction that destabilizes the channel (see Figure 65-3). To test this hypothesis, Oda et al generated a synthetic peptide, DPc10, encompassing the Gly2460-Pro2495 region of RyR2 that mimics an RyR2 mutant: The peptide destabilizes the N-terminal–central region interaction through competitive activity and reproduces the biophysical effect of CPVT mutations.37 The same group engineered several other peptides, mimicking mutations located either in the N-terminal or in the central region of the protein, demonstrating that the mutations caused Ca2+ leakage through the disruption of interdomain interaction.38 Independent support to the hypothesis of unzipping has been provided by George et al by means of high-resolution confocal microscopy and fluorescence resonance energy transfer analysis.39
Store Overload–Induced Calcium Release
The open-state probability of RyR2 increases as the SR Ca2+ increases. If SR Ca2+ reaches a critical threshold, spontaneous Ca2+ leakage may occur even in the absence of membrane depolarization (i.e., calcium-induced calcium release). Chen et al proposed the hypothesis that some RyR2 mutants increase channel sensitivity to luminal Ca2+ (in other words, reduce the threshold for spontaneous Ca2+ release) (see Figure 65-3).25,26,31 This phenomenon was termed store overload–induced calcium release (SOICR). A reduced SOCIR threshold was shown for nine RyR2 mutations. Interestingly, while luminal (i.e., inside the SR) Ca2+ sensitivity was increased, no evidence for an increased sensitivity to cytosolic Ca2+ was observed. On the contrary, Thomas et al reported increased sensitivity of the receptor to cytosolic Ca2+ in L433P and R176Q/T2504M RyR2 mutants.40 While the role of cytosolic Ca2+ sensitivity has still to be determined, it appears evident that a reduced threshold for release has an important role, since, as shown by Venetucci et al, increasing ryanodine receptor open probability alone does not produce arrhythmogenic diastolic Ca2+ release because of the accompanying decrease of SR Ca2+content (and reduced driving force).41,42
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


