Chapter 63 Genetic Diseases
Brugada Syndrome
Sudden cardiac death (SCD) is a major cause of mortality in the Western world, with an approximate incidence of 1 per 1000 per year. Coronary artery disease is the most common cause of SCD. In the absence of coronary disease, SCD is commonly caused by a ventricular arrhythmia in patients with some form of structural heart disease. However, in 10% to 20% of cases, no cardiac structural abnormalities are found at autopsy or after extensive medical investigation of the survivors. In 1992, Brugada and Brugada described a new syndrome causing SCD in individuals with normal hearts.1 Today, generally known as Brugada syndrome, this new entity is thought to be responsible for up to 20% of SCDs that occur in individuals with structurally normal hearts.2
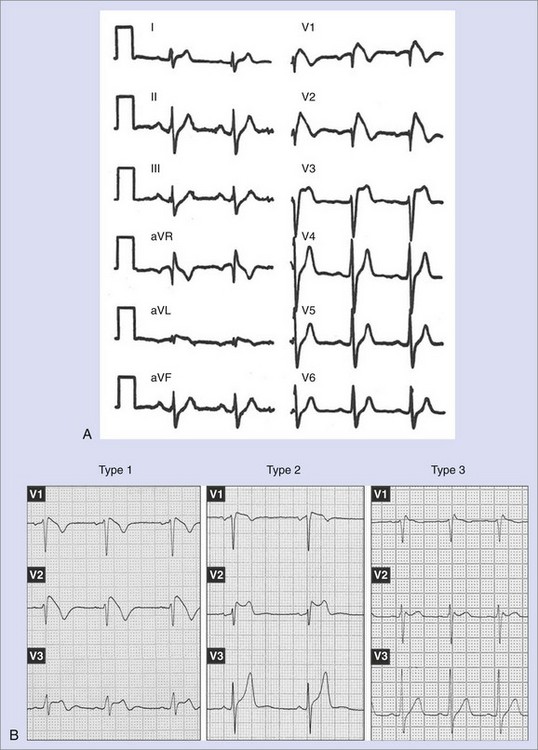
Brugada syndrome was initially described as a clinical syndrome characterized by (1) an electrocardiogram (ECG) resembling a right bundle branch block with a particular morphology of ST-segment elevation in right precordial leads (Figure 63-1, A) and (2) a susceptibility to developing polymorphic ventricular arrhythmias that cause syncope when self-terminating and SCD when long lasting and not terminated by cardiopulmonary resuscitation.1 After the initial description of the first eight patients, numerous works appeared either focusing on clinical characteristics of larger populations or defining the genetic, molecular, and cellular aspects of the disease.3–12 In recent years, major advances in clinical and mechanistic knowledge have provided very valuable information about the disease, but questions still remain, propelling large research activity on the subject. This chapter reviews the current knowledge on clinical, genetic, and molecular features of Brugada syndrome and provides updated information supplied by recent clinical and basic studies.
Definition and Epidemiology
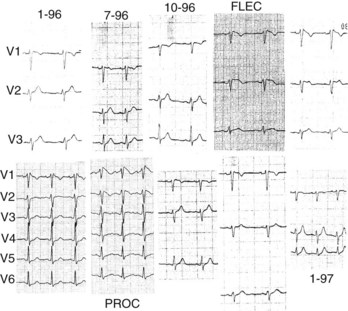
The diagnosis of Brugada syndrome is obvious when the ECG has a certain appearance, as in Figure 63-1, A. However, certain ambiguities appeared in the years following the initial description of the syndrome. Three repolarization patterns were soon identified (Figure 63-1, B)13: (1) type 1 ECG pattern, as described in the initial report in 1992, in which a coved ST-segment elevation 2 mm or greater is followed by a negative T wave, with little or no isoelectric separation, this feature being present in more than one right precordial lead (from V1 to V3); (2) type 2 ECG pattern, also characterized by an ST-segment elevation but followed by a positive or biphasic T wave, which results in a saddle-back configuration; (3) type 3 ECG pattern, a right precordial ST-segment elevation 1 mm or less either with a coved-type or a saddle-back morphology. Although all three patterns can be present in Brugada syndrome patients (see section on Electrocardiography and Modulating Factors), only the type 1 ECG is diagnostic of the syndrome, as it was stated in the first consensus report of the Arrhythmia Working Group of the European Society of Cardiology and subsequently confirmed in the II Consensus Conference published in 2005.2,13 Both documents also held that in order to establish the definite diagnosis of Brugada syndrome, the type 1 ECG pattern should be documented in combination with one of the following clinical criteria: (1) documented ventricular fibrillation (VF), (2) polymorphic ventricular tachycardia (VT), (3) a family history of sudden death (SD) at an age younger than 45 years, (4) the presence of coved-type ECG in family members, (5) inducibility of ventricular arrhythmias with programmed electrical stimulation, (6) syncope, or (7) nocturnal agonal respiration.2,13 However, this definition should be applied with caution, especially when causative mutations have been identified and the disorder can be understood as a disease rather than a syndrome.14,15 In this regard, data from the authors of this chapter confirm that only the presence of the characteristic type 1 ECG pattern, even with no further clinical criteria, may be associated with SD in the follow-up.15 This confirms the need for monitoring all patients, even when an isolated type 1 ECG pattern is found.
The prevalence of Brugada syndrome has been estimated as 5 in 10,000, although this rate may be an underestimation of the real prevalence, as many patients have concealed forms of the disease and remain underdiagnosed. Importantly, significant ethnic and geographic differences have been described.2 For example, the type 1 ECG pattern was observed in 12 of 10,000 in Japan, whereas the few available data on North American and European populations point to a much lower prevalence.16–18 In fact, the syndrome is considered endemic in certain Southeast Asian countries, where it has long been known as sudden unexplained death syndrome (SUDS), also called bangungot (in the Philippines), pokkuri (in Japan), or lai tai (in Thailand); all of these are phenotypically, genetically, and functionally identical to Brugada syndrome.19
Genetics of Brugada Syndrome
Inheritance in Brugada syndrome occurs via an autosomal dominant mode of transmission, although in some cases, the disease can be sporadic, that is, absent in parents and other relatives.2 Thus far, all the mutations that have been linked to Brugada syndrome affect (directly or indirectly) the normal function of specific ion channels participating in the action potential (AP). Therefore, Brugada syndrome is included among the so-called channelopathies, together with other primary electrical disorders such as long QT syndrome (LQTS).
The first mutations related to Brugada syndrome were described in 1998 by Chen and coworkers and were identified in SCN5A, the gene encoding the α-subunit of the cardiac sodium channel (locus 3p21, 28 exons).8 To date, more than 100 other different mutations associated with the syndrome have been found in the same gene.9,12,19–23 For the majority of them, functional studies performed with expression systems have demonstrated a loss of function of the sodium channel that translates into a decrease in sodium current (INa), which is achieved either through a quantitative decrease (failure in their expression) or through a qualitative dysfunction (impaired kinetics) of the channels.9,12,19–23
Although for almost a decade SCN5A has been the only gene linked to Brugada syndrome, mutations in SCN5A are generally found in only 18% to 30% of patients, which suggests a genetic heterogeneity of the disease.2 Accordingly, in the past 2 years, four other genes have been found to be linked to Brugada syndrome. The first of them, glycerol-3-phosphate dehydrogenase 1–like (GPD1-L), was described in 2007 after previous identification of the locus on chromosome 3 (3p22-p24) in 2002.24,25 The A280V mutation in GPD1-L was shown to induce a sodium loss-of-function effect by affecting the trafficking of the cardiac sodium channel to the cell surface.24 Very interestingly, two recent reports demonstrated that mutations in genes other than those involved in the sodium channel function can be responsible for some cases of Brugada syndrome. In 2007, Antzelevitch et al linked loss-of-function mutations in the genes encoding the cardiac calcium channel Cav1.2 (CACNA1c) and its β-subunit CACNB2b to a syndrome overlapping short QT and the Brugada ECG pattern.26 More recently, the same group of investigators described the first family with Brugada syndrome identified to be carrying a mutation (R99H) in the KCNE3 gene, which encodes a β-subunit that is thought to modulate Kv4.3 channels and to be responsible for an increase in transient potassium Ito currents.27 Together, these findings open up new lines of research, where the concept of Brugada syndrome as a pure sodium channelopathy has given way to the concept of the syndrome as an ionic imbalance between inward and outward currents during phase 1 of the AP.
Cellular and Ionic Mechanisms
Experimental studies have elucidated cellular and molecular bases for the two main characteristic features of Brugada syndrome: (1) the specific ECG morphology (ST-segment elevation in right precordial leads) and (2) the susceptibility for VF and SD. Figure 63-2, A, represents the normal ventricular myocyte AP and the major ionic currents involved in each one of the phases. Sodium loss-of-function conditions, the most encountered disorder in SCN5A mutations related to Brugada syndrome, create an imbalance between outward and inward positive currents during phase 1, favoring repolarization and the appearance of a particular notch in the AP (dashed line) that is mediated by the outward transient potassium currents (Ito).9,12,19–23 Comparable imbalances can appear either by decrease in ICaL (in calcium channel loss-of-function mutations) or relative increase in Ito (in the recently described KCNE3 mutation).26,27
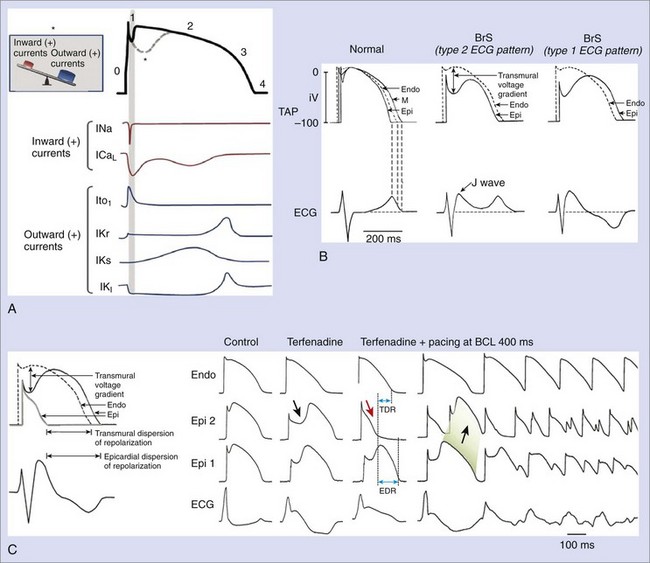
The accentuated notch present in patients with Brugada syndrome, especially in the epicardium, gives rise to a transmural voltage gradient between the epicardium and the endocardium, producing the characteristic ST-segment elevation on ECG (Figure 63-2, B).28 The imbalance between outward and inward positive currents during phase 1 also establishes the basis for the development of ventricular arrhythmias in Brugada syndrome. The proposed mechanism would be a phase 2 re-entry (Figure 63-2, C). When the notch is such that phase 1 reaches approximately −30 mV, all-or-none repolarization can lead to a complete loss of the AP dome. The heterogeneity of the loss of the dome among different sites within the epicardium and between the epicardium and the endocardium results in epicardial and transmural dispersions of repolarization, respectively. This substrate may facilitate the development of premature beats by means of conduction of the AP dome from the sites where it is maintained to the sites where it is lost.11,28
The understanding that the imbalance between inward and outward ionic currents at phase 1 defines the pathologic substrate for Brugada syndrome has multiple implications. First, it has helped in the development of experimental models of the disease, which have been successfully created by the administration of potassium openers (pinacidil), the combination of a sodium channel blocker (flecainide) and acetylcholine, or drugs with combined sodium channel blocker and calcium channel blocker effects.11,29 These interventions create a relative predominance of outward positive currents at the end of phase 1, thus accentuating the notch. The ionic imbalance hypothesis also explains the effects of certain modulators and certain particularities of the syndrome, such as the enhanced phenotypic expression (accompanied by an increased risk of arrhythmias) during vagal situations (acetylcholine inhibits calcium currents, whereas β-adrenergic drugs increase them) or the worse prognosis in men than in women affected with the disease (men could have constitutionally greater Ito density than women).11,30–34 Likewise, it appears that interventions that decrease inward positive currents (as do sodium channel blockers) could be potentially harmful to patients with Brugada syndrome, as they increase ST-segment elevation and the risk of arrhythmic events; however, they could be useful in unmasking concealed forms of the disease.35 In contrast, Ito blockers such as quinidine could be a good therapeutic option, as they reduce the notch at the end of phase 1 (see section on Treatment).36
Clinical Manifestations of Brugada Syndrome
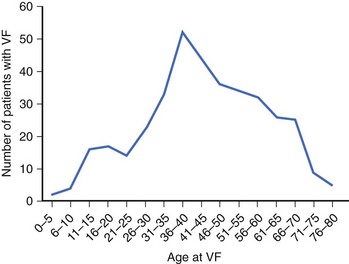
Patients with Brugada syndrome usually remain asymptomatic. However, syncope or cardiac arrest, a consequence of an arrhythmic complication such as polymorphic VT or VF, has been described in up to 17% to 42% of diagnosed individuals.37–40 This rate is probably an overestimation of the real prevalence of symptoms among patients with Brugada syndrome, given that most asymptomatic patients remain underdiagnosed. The age of symptom occurrence (especially cardiac arrest) is consistently around the fourth decade of life in all the series (Figure 63-3), with no definite explanation for this observation thus far.41 Previous syncope may be present in up to 23% of patients who present with cardiac arrest.38 Brugada syndrome should therefore be considered during the workup of patients who have suffered an aborted SD or syncope suspected to be of cardiac origin, especially if no underlying structural heart disease is found (see section on Approach to Patients with Suspected Brugada Syndrome: Family Screening).
Up to 20% of patients with Brugada syndrome may present with supraventricular arrhythmias and thus complain of palpitations, dizziness, or both.42 An increased atrial vulnerability to both spontaneous atrial fibrillation (AF) and induced AF has been reported in patients with Brugada syndrome.43 Other symptoms, such as neurally mediated syncope, have been also recently associated with Brugada syndrome.44,45
As in the case of other sodium channel–related disorders such as type 3 LQTS, ventricular arrhythmias in Brugada syndrome typically occur at rest, especially during night-time or sleep. In a study by Matsuo et al, 26 of 30 episodes of VF documented in implantable cardioverter-defibrillator (ICD) recordings of Brugada syndrome patients appeared during sleep, which suggests that vagal activity may play an important role in the arrhythmogenesis of Brugada syndrome.30 This finding has been confirmed in more recent series.31 As mentioned before, the increase in vagal tone is mediated by acetylcholine decreases in calcium currents, which could favor arrhythmogenesis through a phase 2 re-entry mechanism.11
Sex Differences
It is currently accepted that the clinical phenotype of Brugada syndrome is 8 to 10 times more prevalent in male patients than in female patients.46 Consequently, the main clinical studies published thus far have included 71% to 77% of the male population, which generally appears to be more symptomatic compared with the female population.37–40 The fact that in the case of SUDS, SD mainly occurs in young males during sleep has long been recognized in Southeast Asia, where men from certain small villages dress in women’s bedclothes, as they believe that the syndrome is a female spirit searching for young men at night.
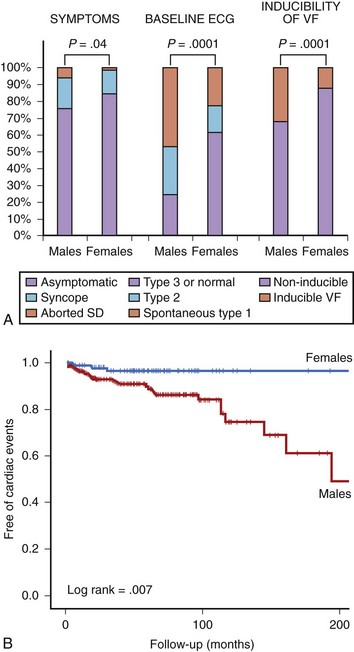
The authors of this chapter recently conducted a study aimed at analyzing gender differences in a large population of patients with Brugada syndrome.33 The study population (n = 384) included 272 men (70.8%) and 112 women (29.2%). General demographic characteristics were similar between both groups (mean age, 45.8 years), but at diagnosis, men presented more frequently with symptoms (syncope in 18%, previous aborted SD in 6%) than did women (14% and 1%, respectively; P = .04). Men also had higher rates of spontaneous type 1 ECG (47% vs. 23%; P = .0001) and inducibility of VF during the electrophysiological study (32% vs. 12%; P = .0001) (Figure 63-4, A).33 The prognosis also differed between men and women. Cardiac events (defined as SD or documented VF) appeared in 31 men (11.6%) and in 3 women (2.8%) during a mean follow-up period of 58 ± 48 months (log-rank test, P = .007). The Kaplan-Meier estimate of cardiac event–free survival according to gender is given in Figure 63-4, B. In accordance with these results, in a recent meta-analysis pooling data from 30 studies and including more than 1500 patients, male gender appeared as an independent predictor of cardiac events defined as SD, syncope, or internal defibrillator shock, with a relative risk (RR) of 3.47 (95% confidence interval [CI], 1.58 to 7.63) compared with women.47
Two main hypotheses have been proposed for the gender distinction, which perhaps interact with each other. First, according to some experimental models, it appears that men could have constitutionally greater Ito density in the right ventricular epicardium than do women, which enhances the ionic imbalance. Second, sex hormones could play a role. Regression of the typical ECG features has been reported in castrated men, and levels of testosterone seem to be higher in male patients with Brugada syndrome compared with those in controls.48,49 Because of this hypothesis, the few data available thus far regarding Brugada syndrome in children have not shown a difference in phenotypic presentation between boys and girls before age 16.50
Children
Although 3 of the 8 patients reported in the first description of the disease were in the pediatric age group, little information has been available on the behavior of Brugada syndrome during childhood.1 Probst et al recently provided data from a multicenter study including 30 patients with Brugada syndrome aged younger than 16 years (mean age, 8 ± 5).50 More than half (n = 17) had been diagnosed during family screening, but symptoms were present in 11 patients (1 aborted SD and 10 syncope). Interestingly, 10 of the 11 symptomatic patients displayed spontaneous type 1 ECG, and in 5 of them, symptoms were precipitated by fever. Five patients received an ICD, and 4 were treated with hydroquinidine.50 During a mean follow-up period of 37 ± 23 months, 3 patients (10% of the population) experienced SD (n = 1) or an appropriate shock from an ICD (n = 2). Importantly, all the 3 patients had presented with syncope at the time of diagnosis and displayed spontaneous type 1 ECG. The 4 patients receiving quinidine remained asymptomatic during 28 ± 24 months of follow-up.50
The results of the chapter authors’ study on 58 pediatric patients with Brugada syndrome are in line with the ones from Probst et al and provide further information on prognosis markers during childhood.51 In the authors’ population (mean age, 11.8 ± 4), up to 22 patients (38%) were symptomatic (11 with previous syncope and 11 with aborted SD), and 36 had been diagnosed during family screening. Spontaneous type 1 ECG was present in 18 patients (31%), and the electrophysiological study (EPS), which was performed in 31 patients, induced VF in 7 of them. An ICD was indicated for 14 patients. Cardiac events appeared in 6 patients (2 SD and 4 appropriate ICD shocks) during a mean follow-up of 48.8 ± 48 months, this rate of 2.5% per year being somewhat lower than the 3.2% per year reported by Probst et al.50 Cardiac events occurred more frequently among patients with spontaneous type 1 ECG and among those with inducible VF at the EPS, but in the authors’ series, symptoms at diagnosis was the strongest variable to predict events during follow-up.51
Though small, these studies clearly suggest the following:
Electrocardiogram and Modulating Factors
As mentioned above, three types of repolarization have been described in patients with Brugada syndrome, but only the coved-type ST-segment elevation (type 1 ECG pattern) is diagnostic of the syndrome (see Figure 63-1). However, it must be stressed that the ECG pattern typically fluctuates over time in patients with Brugada syndrome, and thus can change from type 1 to type 2 or type 3 or even be transiently normal (Figure 63-5). A large variety of conditions influence the electrocardiographic aspect (see below). The prevalence of spontaneous ECG fluctuations was assessed in a work by Veltmann et al, which included 310 ECGs on 43 patients followed up for 17.7 months.52 Among 15 patients with initial diagnostic ECGs, 14 revealed at least one nondiagnostic ECG in a median period of 12 days, while 8 of 28 patients with nondiagnostic ECGs developed a type 1 ECG pattern in a median period of 16 days. On the basis of these results, it appears that repetitive ECG recordings may be mandatory in patients with the syndrome; however, the role of the basal ECG as a risk marker should be understood with some caution (see section on Prognosis and Risk Stratification).52,53
It is worth noting that some factors can account for an ECG abnormality that can closely resemble the Brugada ECG (Table 63-1). Importantly, some of them are conditions different from the syndrome and should be carefully excluded during differential diagnosis, and some others may induce ST-segment elevation probably because an underlying genetic predisposition is present.54
Table 63-1 Electrocardiogram Abnormalities That Can Lead to ST-Segment Elevation in Leads V1 to V3
| DIFFERENTIAL DIAGNOSIS | GENETIC PREDISPOSITION? |
|---|---|
| Atypical right bundle branch block Acute MI, especially of the RV Acute pericarditis/myopericarditis Hemopericardium Pulmonary embolism Dissecting aortic aneurysm Central and autonomic nervous system disorders Duchenne muscular dystrophy Friedreich ataxia LV hypertrophy Arrhythmogenic RV cardiomyopathy Mechanical compression of the RV outflow tract: Mediastinal tumor Pectus excavatum After electrical cardioversion Early repolarization, especially in athletes Hypothermia | Hyperkalemia Hypercalcemia Cocaine intoxication/alcohol intoxication Treatment with: I.Antiarrhythmic drugs: Sodium channel blockers (class IC, class IA) Calcium channel blockers β-blockers II.Antianginal drugs: Calcium channel blockers Nitrates III. Psychotropic drugs: Tricyclic antidepressants Tetracyclic antidepressants Phenothiazines Selective serotonin reuptake inhibitors Lithium |
MI, Myocardial infarction; RV, right ventricle; LV, left ventricle.
From Benito B, Brugada R, Brugada J, Brugada P: Brugada syndrome, Prog Cardiovasc Dis 51(1):1–22, 2008.
Modulating factors play a major role in the dynamic nature of the ECG and also may be responsible for the ST-segment elevation in genetically predisposed patients (see Table 63-1). As mentioned before, sympathovagal balance, hormones, metabolic factors, and pharmacologic agents, by means of specific effects on transmembrane ionic currents, are thought to not only modulate the ECG morphology but also explain the development of ventricular arrhythmias under certain conditions (see section on Cellular and Ionic Mechanisms).11,30,31,48,49,55,56 Temperature may be an important modulator in some patients with Brugada syndrome. Premature inactivation of the sodium channel has been shown to be accentuated at higher temperatures in some SCN5A mutations.10 Accordingly, several case reports in which fever precipitates the syndrome or an arrhythmic complication have been published recently.57,58 It seems that fever would be a particularly important trigger factor among pediatric patients.50
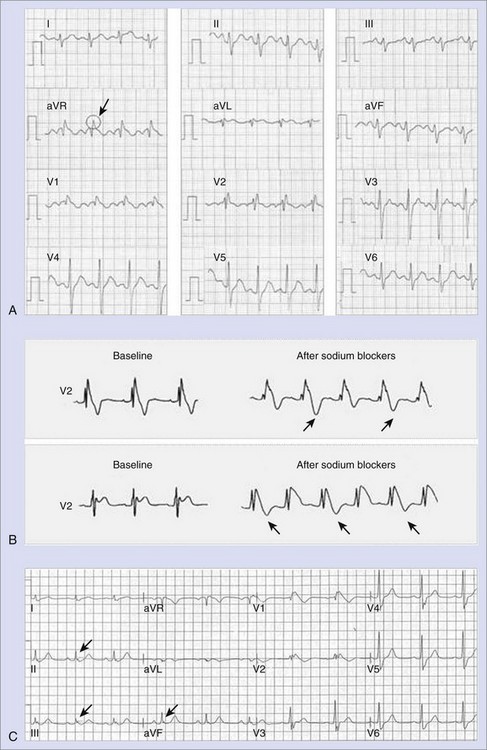
Numerous studies have analyzed the ECG of Brugada syndrome aiming to identify new electrocardiographic hallmarks or risk markers. Pitzalis and coworkers described a prolongation of the corrected Q-T interval (QTc) in right precordial leads, but not in left precordial leads, after administration of flecainide to patients with Brugada syndrome and nondiagnostic basal ECG.59 Subsequently, other groups have correlated a QTc 460 ms or greater in V2 to the occurrence of life-threatening arrhythmias.60 More recently, the aVR sign (an R wave ≥3 mm or an R/q ratio ≥0.75 in lead aVR; Figure 63-6, A) has also been defined as a risk marker of cardiac events in Brugada syndrome, the prominent R wave possibly reflecting some degree of right ventricular conduction delay and consequently more electrical heterogeneity.61 In addition, T-wave alternans (Figure 63-6, B), an indicator of transmural dispersion of repolarization, has been reported after administration of sodium blockers to patients with Brugada syndrome and is thought to be associated with an increased risk for the development of VF.62 Indeed, in a recent study conducted in 77 patients with Brugada syndrome, who were undergoing a pharmacologic test, the presence of T-wave alternans after administration of a sodium blocker identified a subgroup of patients with higher risk of spontaneous VF (52.9% vs. 8.3%, P < .001).63
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


