21
Future developments
Cholesterol-lowering and antiplatelet therapy
Novel cholesterol-lowering secondary prevention therapies
Increasing high-density lipoprotein (HDL)
Multiple large-scale epidemiological studies have consistently shown an inverse relationship between HDL cholesterol levels and cardiovascular risk. Studies have shown that for every 0.03 mmol/L increase in HDL there is an associated 2–3% reduction in cardiovascular risk.1 HDL mediates its cardioprotective role through several mechanisms. One of these is the ability of HDL to remove macrophage cholesterol through its involvement in reverse cholesterol transport (Fig. 21.1). It transports excess cholesterol from the arterial macrophages to the liver, where it can be excreted in the bile. HDL also has direct anti-inflammatory and antithrombotic effects that have been attributed to the action of apolipoprotein A-I, a protein derived from HDL that will be discussed below (Fig. 21.2).
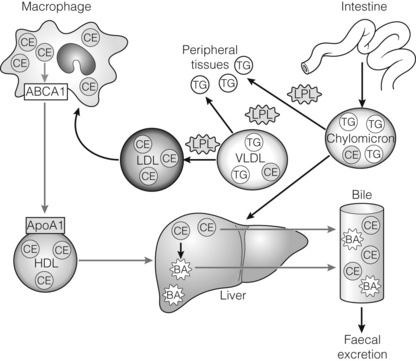
Figure 21.1 Overview of lipid transport and metabolism. Dietary lipids in the form of triglycerides (TG) and cholesterol esters (CE) are absorbed through the intestinal wall and packaged as chylomicrons. These triglyceride-rich particles are rapidly metabolised by lipoprotein lipase (LPL), which hydrolyses triglycerides into free fatty acids and glycerol that are subsequently taken up by peripheral tissues. Chylomicron remnants are removed from circulation by the liver. The liver synthesises triglyceride-rich very-low-density lipoprotein (VLDL) particles as a means of transport for de novo cholesterol and triglycerides. VLDL particles can also be converted to low-density lipoproteins (LDL) through the actions of LPL and function to deliver cholesterol to peripheral tissues such as macrophages. Small phospholipid-rich high-density lipoprotein (HDL) can be made by the liver or from pinched-off VLDL or chylomicron remnants. HDL functions in a process known as reverse cholesterol transport (RCT; red arrows). RCT involves HDL-mediated removal of excess cholesterol from peripheral tissues through the interactions of apolipoprotein A-I (ApoAI) on the surface of HDL and ABCAI on the surface of peripheral cells. This cholesterol is transported to the liver in the form of HDL, where it is converted to bile acids (BA) or secreted directly into the bile. Both bile acids and cholesterol present in the bile can be removed from the body through excretion in the faeces.2 Reproduced from Hanniman EA, Sinal CJ. Nuclear receptors: novel therapeutic targets for the treatment and prevention of atherosclerosis. Drug Discovery Today: Therapeutic Strategies 2004; 1:155–61. With permission from Elsevier.
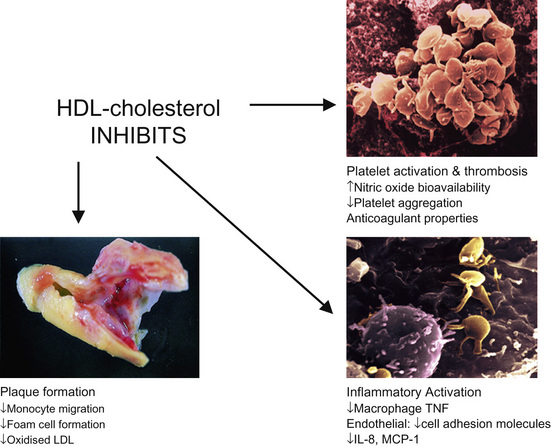
Figure 21.2 Anti-inflammatory properties of HDL. IL, interleukin; MCP-1, monocyte chemoattractant protein 1; TNF, tumour necrosis factor.3
It should be noted that HDL particles and apolipoproteins which are dysfunctional and exhibit pro-inflammatory properties have been identified in some patient cohorts.4 Thus the functionality as well as the quantity of HDL cholesterol that is present is important.
Cholesterol ester transfer protein inhibitors: Various pharmacotherapies have aimed to increase HDL levels. Recent interest has focused on the use of inhibitors of cholesterol ester transfer protein (CETP). In a randomised double-blind study of 15 000 patients with a history of cardiovascular disease, including PAD, the CETP inhibitor torcetrapib was associated with increased risk of cardiovascular events and mortality.5 Torcetrapib when used in patients already on atorvastatin resulted in a 72% increase in HDL cholesterol and a 25% reduction in low-density lipoprotein (LDL) cholesterol. However, the increased HDL generated by torcetrapib was shown to be less effective with regard to reverse cholesterol transport. Torcetrapib reduced serum potassium and increased bicarbonate levels, aldosterone levels and blood pressure. These effects were not related to CETP inhibition and further work led to the development of the more specific CETP inhibitors: anacetrapib, dalcetrapib and evacetrapib, which have been evaluated in phase I trials.6
The safety and efficacy of anacetrapib in increasing HDL cholesterol has been assessed in a randomised placebo-controlled double-blind study involving 1623 patients with cardiac disease or high risk of cardiac disease.7 In the Randomised Evaluation of the Effects of Anacetrapib through Lipid Modification study (REVEAL), patients at high risk of vascular events, which includes patients with PAD, are currently being recruited into a large multicentre trial with the aim of determining if anacetrapib prevents vascular events.8 The patients will undergo optimisation of LDL cholesterol via atorvastatin therapy prior to entering the study and will be randomised to receive anacetrapib 100 mg daily or placebo for 5 years.
Niacin: Niacin is known to increase HDL cholesterol levels, but the ability to reduce cardiovascular events in patients on statin therapy is unknown. One large randomised trial of niacin monotherapy was performed before the introduction of statin therapy.9 Niacin causes flushing, which may be reduced by laropiprant (formerly MK-0524), a selective prostaglandin D receptor antagonist. In the Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) randomised placebo-controlled study, 25 000 patients with atherosclerotic vascular disease including PAD have been randomised to niacin (1 g)/laropiprant in addition to pre-existing simvastatin or combination simvastain/ezetimibe therapy for 4 years.10 Prior to entry into the study the patients’ LDL cholesterol level was below 2 mmol/L. The aim is to determine if increasing HDL cholesterol levels through niacin therapy reduces the primary outcome of myocardial infarction, stroke or revascularisation.
In the study of niacin in patients with low HDL cholesterol levels receiving intensive statin therapy (AIM-HIGH), 3414 patients with atherosclerosis were randomised to extended-release niacin (1.5–2 g daily) or matching placebo.11 All patients had an LDL cholesterol level of 1.03–2.07 mmol/L and were on simvastatin plus ezetimibe if required. The primary end-point was the first event of a composite, myocardial infarction, stroke, hospitalisation for an acute coronary syndrome, revascularisation or death from coronary heart disease. The study was stopped after a mean follow-up period of 3 years since due to a lack of efficacy, as despite significant increases in HDL-cholesterol there was no observed increase in clinical benefit.
Apolipoprotein A-I and mimetic peptides: Apolipoprotein A-I is a gatekeeper in reverse cholesterol transport and binds to the ABCA1 receptor on macrophages (Fig. 21.1). Apolipoprotein A-I has been shown in several preclinical studies to reduce the development and progression of atherosclerosis through its lipid-binding properties. It is, however, a large protein that has to be administered intravenously. Thus, D-4 F peptide has been developed, which appears to mimic the lipid-binding actions of apolipoprotein A-I and can be administered orally.12 Preclinical trials have shown that D-4 F has anti-inflammatory and anti-atherogenic effects. In a small phase I clinical trial, D-4 F had low bioavailability but a single dose appeared safe and well tolerated.13 Further studies are awaited.
Summary: therapy to increase levels of HDL cholesterol: While there is robust epidemiological evidence that low levels of HDL cholesterol are associated with increased risk of cardiac events, to date there is no evidence that therapy which aims to increase HDL cholesterol levels reduces this risk. In the era of aggressive statin therapy, which can achieve very low LDL cholesterol levels, the clinical benefit of improving HDL cholesterol levels remains to be proven.
Lipoprotein metabolism and inflammatory response
The role of lipids in the pathogenesis of atherosclerosis has been discussed in Chapter 1.
Secretory phospholipase A2 and lipoprotein-associated phospholipase A2 are two inflammatory biomarkers that have been shown to predict cardiovascular events in population-based studies of healthy volunteers and in patients with known coronary heart disease.14 They have not been studied in patients with PAD.
Secretory phospholipase A2 causes hydrolysis of phospholipids on lipoproteins and cell membranes, and the generation of pro-inflammatory lipids (non-esterified fatty acids, lysophospholipids and eicosanoids). Lipoprotein-associated phospholipase A2 generates pro-inflammatory lipids from oxidised LDL (oxidised non-esterified fatty acids and lysophosphatidylcholine).15
Secretory phospholipase A2 inhibitors: The secretory phospholipase A2 inhibitor, varespladib methyl, has been shown in two large phase II studies to significantly reduce the levels of LDL cholesterol, non-HDL cholesterol and ApoB.16,17 A phase III study involving 6500 patients with acute coronary syndrome is currently underway.18
Lipoprotein-associated phospholipase A2 inhibitors: The lipoprotein-associated phospholipase A2 inhibitor darapladib has been evaluated in two phase II studies.19,20 Darapladib reduced lipoprotein phospholipase activity and concentrations both within the blood and in carotid plaques. The concentrations of other inflammatory mediators were also reduced but there was no change in lipid profiles. Currently, there are two phase III studies underway that aim to determine the effect of darapladib on clinical outcome in patients with a recent cardiovascular event and patients with stable disease.21,22 The ability of these drugs in combination with statin therapy to improve clinical outcome needs to be determined.
Novel antiplatelet agents
The value of antiplatelet therapy in patients with PAD has been discussed in Chapter 1. The three major pathways involved in platelet activation are triggered by thromboxane, adenosine diphosphate and thrombin (Fig. 21.3).
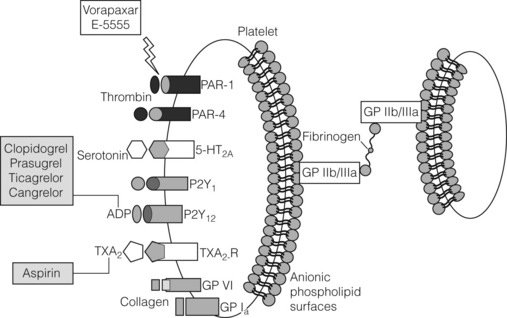
While antiplatelet therapy has been effective at reducing thrombotic risk in patients with atherosclerosis, patients continue to experience thrombotic events. Many of these events occur because patients may be non-responsive to current antiplatelet therapy. For instance, the thienopyridine clopidogrel needs to be activated by cytochrome P450 metabolism, and recent studies have shown that clopidogrel is inactive in patients who lack a functional allele of CYP2C19.23 A newer generation of P2Y12 receptor antagonists and novel agents such as thrombin receptor antagonists have recently been developed and are discussed below.
Thrombin receptor antagonists: protease-activated receptor 1 (PAR-1)
Platelet thrombin receptor antagonists, also known as protease-activated receptor 1 (PAR-1) antagonists, are a novel class of antiplatelet agents.24 In phase II clinical trials, two oral PAR-1 antagonists, vorapaxar (SCH 530348, Merck/Schering-Plough) and atopaxar (E-5555, Eisai Co), have been evaluated in combination with standard antiplatelet therapy. In the thrombin receptor antagonist trial of patients undergoing percutaneous coronary angioplasty (TRA-PCI), vorapaxar was found to be safe and effectively inhibited platelet function in a thousand patients.25 Results of the phase II trials involving atopaxar are awaited.26
Vorapaxar: phase III trials
In the phase III TRACER study (Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome), 12 944 patients with acute coronary syndrome without ST-elevation were entered into a double-blind randomised controlled trial of vorapaxar. The primary end-point was a composite of myocardial infarction, stroke, and death from cardiovascular causes, recurrent ischaemia or urgent coronary revascularisation. The study was terminated early after 6473 patients were randomised, due to the increased risk of major bleeding in patients randomised to vorapaxar, which included intracranial haemorrhage. The study failed to show any difference in its composite end-points.27
However, in the phase III TRA-2P (Thrombin Receptor Antagonist in Secondary Prevention of atherothrombotic ischaemic events) study, 26 449 patients with a prior history of myocardial infarction, ischaemic stroke or symptomatic PAD were randomised to vorapaxar or placebo in addition to standard care.28 Patients randomised to vorapaxar had a significantly reduced risk of sustaining the primary end-point, which was a composite of cardiovascular death, myocardial infarction, stroke or urgent coronary revascularisation (hazard ratio 0.88; 95% confidence interval (CI) 0.82–0.95; P = 0.001). This, however, was associated with an increased risk or moderate and severe bleeding complications. In particular, as in the TRACER study, a significantly increased risk of bleeding, including intracranial haemorrhage (ICH), was observed in patients randomised to vorapaxar.
P2Y12 receptor antagonists
Prasugrel is a third-generation thienopyridine, which is more potent, has a faster onset of action and, unlike clopidogrel, its bioconversion is not affected by CYP genetic polymorphisms.29 In a phase III trial, 3534 patients with ST-elevation myocardial infarction who were due to undergo percutaneous coronary intervention were randomised into a placebo-controlled double-blind trial (TRITON-TIMI38).30 Prasugrel was more effective than clopidogrel in reducing the primary end-point of cardiovascular death, non-fatal myocardial infarction or stroke. The major bleeding rates were similar apart from increased bleeding after coronary artery bypass graft (CABG) surgery.
Ticagrelor is the first oral reversible P2Y12 antagonist, it is a cyclopentyltriazoloprimidine which does not require metabolic activation and may be of value in patients who require to undergo surgery. In the PLatelet Inhibition and Patient Outcomes (PLATO) trial, ticagrelor reduced the incidence of death as a result of cardiovascular causes with no increase in overall major bleeding or bleeding related to CABG compared with clopidogrel. However, it was associated with increased non-CABG major bleeding and non-procedural-related major bleeding after 30 days on study treatment.31 Ticagrelor was also associated with increased side-effects such as dyspnoea, hypotension and arrhythmias (ventricular pauses). Cangrelor is an intravenous P2Y12 antagonist with a very fast onset and short half-life. It may have benefit in patients undergoing cardiac surgery.32 Further studies are awaited.
EP3 receptor antagonists
Production of prostaglandin E2 by an inflamed plaque potentiates atherothrombosis. Similar to the P2Y12 receptor, the platelet EP3 receptor for prostaglandlin E2 also inhibits cyclic AMP synthesis, thus resulting in platelet activation. EP3 receptor antagonists such as DG-041 may have the potential to selectively reduce atherothrombosis without increasing the risk of bleeding and are currently being evaluated in clinical trials.33,34
Angiogenesis
Therapeutic angiogenesis aims to stimulate the development of new vessels from the pre-existing vasculature in order to bypass occluded segments.35,36 Angiogenesis is a complex process involving many growth factors interacting to produce the end-result of neovascularisation and reversal of ischaemia. The natural trigger for angiogenesis is hypoxia. Hypoxia induces expression of a variety of growth factors, including vascular endothelial growth factor (VEGF) and angiopoietin-2 (Ang-2). These act on cells located within the basement membrane of capillaries known as pericytes, which play an important role in endothelial cell proliferation, migration and stabilisation. A number of phases are involved in the angiogenesis process (Fig. 21.4). In the initiation phase, pericytes detach from the capillaries, basement membrane is degraded by matrix metalloproteinases (MMPs) and plasma proteins extravasate to form a protein-rich matrix in the interstitial space. In the neovascular phase, growth factors up-regulated by hypoxia and released from the extracellular matrix induce proliferation and migration of endothelial cells and pericytes. In the adaptation phase, excessive neovessel network is then trimmed to respond to the metabolic needs of the tissue and vessels that receive insufficient blood flow regress. In the maturation phase, vessels with sufficient flow mature, achieve pericyte coverage and deposit a basement membrane.37 VEGF is the most potent angiogenic factor and is the most studied gene in trials to date.38 Other pertinent factors and genes that have been studied are fibroblast growth factor (FGF), hypoxia-inducible factor-1 alpha (HIF-1α), hepatocyte growth factor (HGF) and developmental endothelial locus-1 (Del-1).
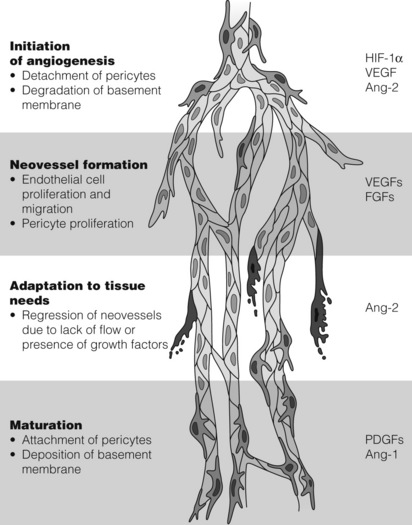
Figure 21.4 Angiogenic growth factors and their effect on the angiogenesis pathway. Ang-1, angiopoietin-1; Ang-2, angiopoietin-2; FGF, fibroblast growth factor; HIF-1α, hypoxia-inducible factor-1 alpha; PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor. Reproduced from Markkanen JE, Rissanen TT, Kivela A et al. Growth factor-induced therapeutic angiogenesis and arteriogenesis in the heart–gene therapy. Cardiovasc Res 2005; 65:656–64. With permission from Oxford University Press.
Target genes in use for gene therapy in peripheral arterial disease
A highly specific mitogen for endothelial cells, VEGF is a potent initiator of angiogenesis. Its signal transduction results in endothelial cell proliferation, migration and ultimately new vessel formation.38 These pro-angiogenic cytokines are a multigene family, of which VEGF-A is the most commonly studied, particularly VEGF-A121 and VEGF-A165, as these are associated strongly with new vessel formation.39 Similarly, FGF is a family of heparin-binding polypeptide proteins that are powerful stimulators of angiogenesis. They control the proliferation of endothelial cells, smooth muscle cells and fibroblasts, and the most widely studied FGF, FGF-1, has been shown to play a key role in neoangiogenesis in preclinical trials in vivo.40,41 A regulator of angiogenesis, HIF-1α, plays a vital role in the cellular response to hypoxia. In the presence of low oxygen tensions, this factor is released and stimulates the production of several pro-angiogenic growth factors, including VEGF-A.42 Hepatocyte growth factor is a pro-angiogenic growth factor that has a predilection for the stimulation and proliferation of epithelial and endothelial cells.43 Del-1 is expressed widely during vascular development in the embryonic period. It has the role of encouraging endothelial cell adhesion and preclinical studies have proven its presence in ischaemic tissue, suggesting it has a role as an intermediary of neoangiogenesis44,45 (Fig. 21.4).
Gene therapy vectors
Non-viral methods
Non-viral methods for stimulating angiogenesis make use of naked plasmid DNA. Several advantages are associated with the use of plasmids: ease of production, high yields and, owing to its low immunogenic potential, its safety profile.46 Despite these advantages, large randomised trials have shown that gene transfer efficiency is low, limiting its overall effect.47 This has led to a greater focus in developing viral vectors as the method of choice.
Viral methods
Viruses, as part of their replication cycle, will enter the cell and integrate their genetic information with the DNA of the native cell, with subsequent expression of the desired effects. However, they initiate an immune response and older viral types were unable to integrate with cells in the quiescent phase of the cell cycle, which are found abundantly in vascular tissues.48 The best known and most widely used viral vectors studied come from the family of adenoviruses.49 These vectors have the associated advantages of: low mutagenesis (as they do not integrate with the host genome); high titres; broad tissue tropism; and ability to induce cells in the quiescent phase.50 Stimulation of the immune response is, however, unavoidable and this acts to decrease their effectiveness, especially after repeated administrations, accounting for the short-term improvements with poorer longer-term outcomes.51
Delivery methods of vectors
The delivery of viral vectors can be directly into the circulation at the time of surgery or endovascular techniques, or by the intra-arterial route, or by the intramuscular route in those patients not suitable for any form of endovascular or surgical approach. Direct intra-arterial administration is limited by its transient response as it is distributed to a wide area, while intramuscular injection has the advantage of local penetration to the area of ischaemia with a longer-lasting response.52
Non-viral methods use physical or chemical forces, which are intended to induce cell membrane defects to allow the DNA (in plasmid form) to diffuse and create its effect intracellularly. The physical methods include microinjection, ultrasound and hydrodynamic administration, while chemical approaches use natural or synthetic substances to bind cellular receptors to gain entry into the cell and then interact with the DNA to exert their response. This non-viral method is associated with low transfection rates and transient gene expression. However, its advantages are its ease of use and low immunogenic potential.37
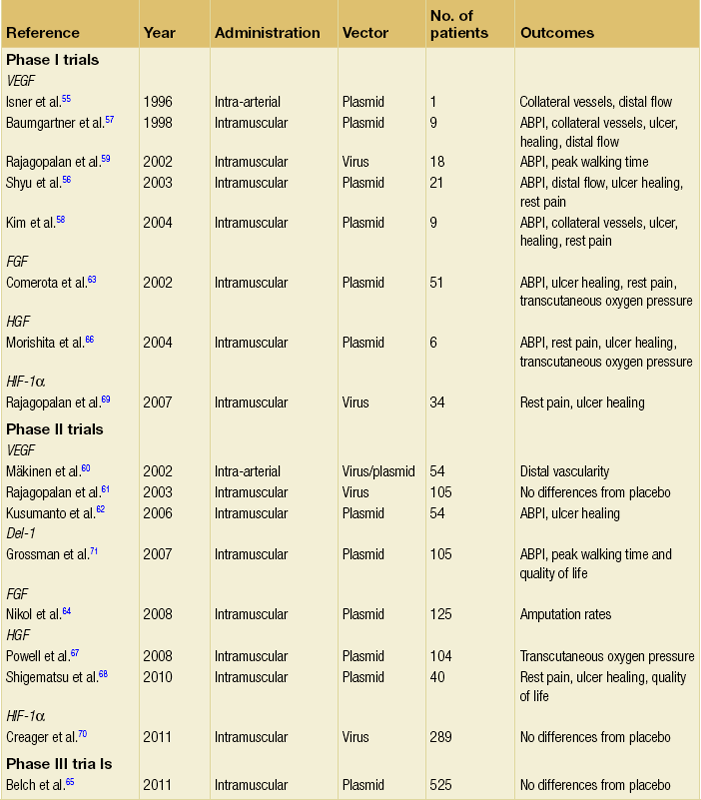
Therapeutic angiogenesis has been used in in vitro models, animal models and clinical trials.53,54 This section will outline the clinical trials that have been conducted and summarise the current issues in the field of gene therapy for PAD (Table 21.1).
Table 21.1
Methodology employed by the trials assessing gene therapy for peripheral arterial disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


