Fibroblasts in Lung Homeostasis and Disease
INTRODUCTION
Fibroblasts are the primary cellular source responsible for synthesis and remodeling of the extracellular matrix (ECM). These cells are in communication with the surrounding microenvironment and play a key role in lung homoeostasis. Following lung injury, fibroblasts are activated and undergo myofibroblast differentiation. Myofibroblasts are key effector cells for lung repair following injury. In addition to fibroblasts, perivascular pericytes and mesenchymal stem cells (MSCs) of bone marrow (BM) origins contribute to myofibroblast population. There is evidence that type II alveolar epithelial cells can differentiate into myofibroblasts in vitro through a process known as epithelial–mesenchymal transition (EMT); however, the role of EMT in fibrogenesis in vivo remains controversial. Myofibroblasts express α-smooth muscle actin (α-SMA), develop robust actin filaments (stress fibers), and acquire contractile activity. The function and behavior of myofibroblasts are regulated by both biochemical and physical cues in the surrounding microenvironment. The fate of myofibroblasts is a key determinant of whether an injury–repair response will resolve or progress into fibrosis. Destruction and aberrant remodeling of the ECM is a common feature of many lung diseases, including pulmonary fibrosis, asthma, chronic obstructive pulmonary disease (COPD), and lung cancer. Targeting myofibroblasts and tissue remodeling may provide a novel and effective strategy for treating a number of chronic lung diseases.
FIBROBLAST BASICS
 WHAT ARE FIBROBLASTS?
WHAT ARE FIBROBLASTS?
Fibroblasts were described as early as in the late 19th century, based on their location and their microscopic appearance.1 These are elongated cells that display a spindle-shaped morphology with extended cell processes.2 Fibroblasts are ubiquitous in tissues and organs throughout the body and communicate with other cells such as epithelial cells (Fig. 28-1). Despite its discovery over a century, a reliable and specific molecular marker that identifies the fibroblast is currently lacking. Many indicators of fibroblast phenotype have been suggested in the previous studies (e.g., fibroblast-specific protein 1, vimentin, prolyl 4-hydroxylase, procollagen-Iα2, etc.).3 However, none of them are specific to fibroblasts and/or are present in all fibroblasts. Currently, fibroblasts are identified by their ability to adhere to plastic, and their lack of markers that indicate other cell lineages. Clearly, better cellular markers with absolute specificity for fibroblasts will aid in the study of sources, differentiation, and phenotypic plasticity of fibroblasts.
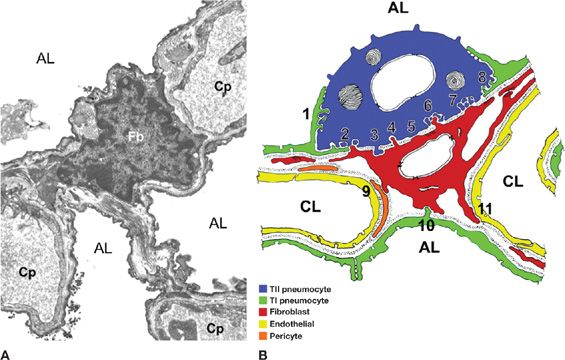
Figure 28-1 An interstitial fibroblast in the alveolar wall. A. A transmission electron microscopic image showing the structural organization of the alveolar wall in canine lung. Fb, fibroblast; Cp, capillary; AL, alveolar lumen; (B) summary of fibroblast (red) relationships with type I (green) and type II (purple) alveolar epithelial cells, capillary endothelial cells (yellow), and pericytes (orange) in human and rabbit alveolar walls. (Reproduced with permission from Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev. 2003;83(2):309–336.)
Tissue-Specific Fibroblasts and Fibroblast Heterogeneity
Fibroblasts isolated from different tissues display a considerable degree of heterogeneity in phenotype and activity.4 Such diversity is evident by divergent and specific gene expression patterns among fibroblasts isolated from distinct anatomical locations.5 For example, fetal skin fibroblasts express high levels of collagen types I and V, whereas fetal lung fibroblasts lack collagen I and V expression.5 Instead, fetal lung fibroblasts exclusively express lung-specific forkhead family transcription factors FOXF1 and FOXP1.5 Tissue-specific fibroblasts may provide location-specific signaling for a given anatomic origin as well as important positional cues for wound healing and tissue regeneration. Besides the differences in fibroblasts from different anatomical sites, fibroblasts derived from a single tissue are often composed of subsets of different fibroblasts.6 For example, fibroblast subpopulations isolated from lung differ in expression of surface markers such as Thy-1, cytoskeletal composition, lipid content, and cytokine profile.7,8 Fibroblasts isolated from lungs with active fibrotic disease such as in the fibroblastic foci of human idiopathic pulmonary fibrosis (IPF) are morphologically and functionally distinct from fibroblasts isolated from normal lungs,9 suggesting that selective expansion of specific fibroblast subsets is associated with the pathogenesis of this disease.
 FIBROBLAST FUNCTIONS
FIBROBLAST FUNCTIONS
The important functions of fibroblasts include deposition of ECM, regulation of inflammation, and wound healing.10 Fibroblasts produce ECM-degrading proteases such as matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs), supporting their crucial roles in regulating ECM turnover and homeostasis.11
ECM Production
One of the major functions of fibroblasts is the production of ECM. The ECM in the lung provides an essential scaffold on which cells can proliferate and differentiate. It also provides the lung with tensile strength and elasticity that are essential for ventilation. Lung ECM is composed of fibrillar proteins, glycoproteins, proteoglycans (PGs), and polysaccharides, each of which has distinct biochemical and biomechanical properties; these include collagens, elastin, fibronectin, PGs, hyaluronan (HA), laminin, vitronectin, and thrombospondin.12 Type I and type III collagens are the most abundant collagens in the lung interstitium (ratio 3–6:1). Collagen IV is mainly localized to basement membranes. A fibroblast is estimated to synthesize approximately 3.5 million procollagen molecules/cell/day.13,14 Depending on tissue type and age, 10% to 90% of synthesized procollagens are intracellularly degraded by lysosomal enzymes (e.g., cathepsins B, D, and L). It is postulated that regulation of procollagen-degrading rate may provide an important mechanism for rapid secretion of collagen in response to injury without de novo synthesis of new proteins.
Secretion of Proteolytic Enzymes and Inhibitors
The ECM is a dynamic structure that undergoes constant remodeling. Remodeling of the ECM is regulated by complex mechanisms including stimulatory and inhibitory mediators derived from resident cells in the local environment.14 Fibroblasts synthesize a variety of proteolytic enzymes and inhibitors that enable them to control the assembly and turnover of the ECM. MMPs are either secreted by fibroblasts as inactive zymogens or anchored to the cell surface. Activation of MMPs occurs by disruption of interactions between the prodomain and the catalytic domain through either the proteolytic cleavage or the conformational change of the proenzymes.15 MMPs function as proteinases that degrade most ECM proteins. Proteolysis of the ECM macromolecules by MMPs results in the release of cryptic fragments and neoepitopes that promote angiogenesis and cellular migration.16,17 MMPs also have critical roles in the posttranslational regulation of other proteins including latent growth factors stored within the ECM, membrane receptors, and other proteases.18 Hence, MMPs impact cell behavior both through modulation of cell–matrix interactions and through regulation of other signaling molecules. MMPs themselves are regulated by their endogenous inhibitors, TIMPs. TIMPs block MMP activity by noncovalently binding to the MMP active site. A tight balance between MMP proteolysis and TIMP expression is required for maintaining lung homeostasis.19
Innate Immune Function
Fibroblasts are capable of synthesizing many inflammatory cytokines that are initially thought to be exclusively produced by inflammatory cells.20 Fibroblast-derived cytokines play an important role in the amplification and perpetuation of the immune response. Fibroblasts generate constitutive and cytokine-induced C-C and C-X-C chemokines that recruit inflammatory and immune cells to the injured cites.21 Lung fibroblast–derived granulocyte macrophage colony–stimulating factor promotes the survival of eosinophils, which contributes to the fibrotic response in the lung.22,23 Direct contacts between fibroblasts and T cells promote the production of adhesion molecules and cytokines by the T cells.24 Interactions between fibroblasts and mast cells facilitate de novo production of eotaxin, a potent eosinophil chemoattractant.25 The impact of interactions between fibroblasts and inflammatory cells are bidirectional. Th2 cells produce IL-4 that recognizes specific receptors on fibroblasts and modulates fibroblast proliferation and biosynthetic capacity.26 Eosinophils release mitogens that augment fibroblast proliferation and collagen production.27,28 Fibroblasts are the main producers of ECM proteins. Since ECM components affect multiple functions and properties of inflammatory and immune cells,29,30 it suggests a further regulatory role of fibroblasts in innate immune response by the effects on the ECM.
Regulation of Tissue Interstitial Fluid Pressure and Microhemodynamics
Fibroblasts regulate tissue interstitial fluid volume, pressure, and microcirculation hemodynamics by generation of actomyosin-derived forces and transmission of the forces to the ECM via transmembrane integrins.31 Such mechanical interactions between fibroblasts and matrix are subjected to modulation by soluble factors, such as PDGF that results in increased interstitial fluid pressure, and TNF-α that leads to decreased interstitial fluid pressure.32,33 Fibroblasts express ecto-5′-nucleotidase (CD73) and soluble guanylyl cyclase (cGC) that affect hemodynamics,34 suggesting a role of fibroblasts in the regulation of the microcirculation.
MYOFIBROBLASTS: EFFECTOR CELLS IN TISSUE REPAIR
 HISTORICAL CONTEXT
HISTORICAL CONTEXT
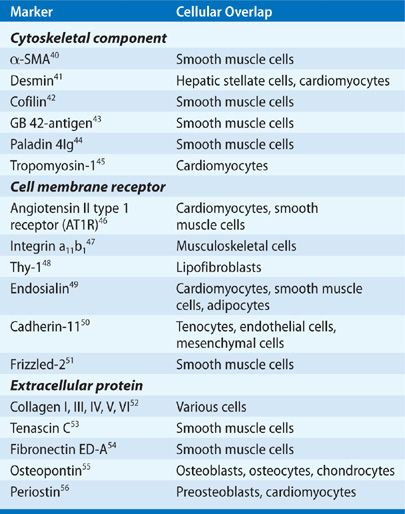
Myofibroblasts were first identified as fibroblastic cells with a strong muscle cell–like contractile filamentous apparatus in the granulation tissue of healing wounds.35 These cells are present in organs with increased remodeling, such as in development, inflammation, fibrosis, cancer invasion, and metastasis.36 Myofibroblasts display prominent cytoplasmic actin microfilaments (stress fibers) (Fig. 28-2). They are connected to one another with adherens and gap junctions as well as to the ECM with focal adhesion (FA) complexes.37 Myofibroblasts may be further classified into subtypes based on the presence or absence of vimentin, desmin, and/or α-SMA in cytoskeletal filaments. Expression of these proteins varies upon tissue types and is subjected to the regulation by environmental factors.38 Alternatively, myofibroblasts can be simply classified into two subpopulations, proto-myofibroblasts and mature myofibroblasts.39 Proto-myofibroblasts are partly differentiated myofibroblasts that contain α-SMA–negative actin stress fibers, whereas mature myofibroblasts possess extensive network of α-SMA–positive stress fibers and large FAs (termed supermature FAs). α-SMA is the most widely used marker for identification of myofibroblasts (Fig. 28-2). In addition, studies have identified several other markers and modulators of myofibroblasts (Table 28-1). However, a specific and universal myofibroblast marker remains to be identified.
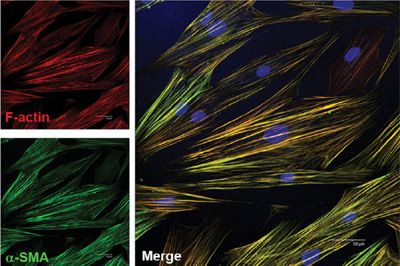
Figure 28-2 Myofibroblasts are characterized by de novo synthesis of α-SMA and incorporation of α-SMA into filamentous actin stress fibers. Scale bar: 50 μm.
TABLE 28-1 Myofibroblast Markers
 ORIGIN OF MYOFIBROBLASTS
ORIGIN OF MYOFIBROBLASTS
The precise origin of myofibroblasts remains unsolved at present. Studies have suggested that local fibroblasts, perivascular pericytes, BM-derived MSCs, tissue MSCs, and epithelial/endothelial cells (through epithelial/endothelial–mesenchymal transition) are among potential cellular sources for myofibroblast population.
Resident Fibroblasts—Mesenchymal Stem Cells
Fibroblasts are the most widely accepted origin for myofibroblasts.57,58 Fibroblasts influx into injured sites from the surrounding tissue and differentiate into myofibroblasts in response to extracellular stimuli including biochemical and physical cues.59 These stimuli induce intrinsic changes in gene expression and stress fiber formation that characterizes myofibroblast differentiation.
MSCs that reside in tissues lack hematopoietic and leukocyte markers, but may express α-SMA.60 Tissue MSCs have been described in the dermal sheath that surrounds the hair follicle facing epithelial stem cells.61 These MSCs are involved in papilla regeneration and differentiate into myofibroblasts in response to environmental insults. It has been reported that MSCs from subcutaneous fat are responsible for collagen accumulation in scars.62 Following lung injury, myofibroblasts were found to originate from perivascular and peribronchial sources.63 It is likely that there are tissue MSCs in the human adult lung,64 which become activated and undergo myofibroblast differentiation in response to lung injury. In support of this notion, a previous study has provided evidence for isolation of postnatal MSCs from different organs including liver, kidney, and lung.65
Pericytes
Pericytes are perivascular cells located on the abluminal side of endothelial cells in microvasculature.66 These cells share developmental origins with fibroblasts, but differ from fibroblasts by the fact that pericytes anatomically connect with endothelial cells through cell processes within capillary basement membrane, whereas fibroblasts do not directly interact with endothelial cells.67 The normal function of pericytes includes the regulation of vascular tone and blood flow through expression of contractile microfilaments (actin, myosin) and intermediate filaments (desmin, vimentin).68 Microvascular pericytes differentiate into myofibroblasts in diffuse cutaneous systemic sclerosis, providing a link between microvascular damage and skin fibrosis.69 Recent studies using the genetic fate-mapping approach have clearly demonstrated that pericytes are a major cellular source of myofibroblasts in animal models of acute injury to muscle, dermis, and kidney.70,71 In addition, HSCs, pericyte-like cells in the liver, are the primary myofibroblast progenitors in mouse models of alcoholic and toxic liver fibrosis.72
Bone Marrow–Derived Mesenchymal Cells
BM-derived MSCs are self-renewable, multipotent progenitor cells with the capacity to differentiate into lineage-specific cells that form bone, cartilage, fat, tendon, and muscle.73 Compared with hematopoietic stem cells, MSCs are more radio-resistant and reside mostly in BM stroma. BM-derived MSCs do not express hematopoietic markers and can be isolated as Lin-CD45-CD31-CD34-CD133-Sca-1+Vitamin A-cells.74 In vitro evidence for a BM origin of myofibroblasts was first presented more than four decades ago. By plating BM cells on culture dishes, colonies consisting of cells exhibiting elongated or polygonal cytoplasm and clear nuclei were formed.75 Transplantation studies using Y chromosome or green fluorescent protein (GFP) as a marker of donor cells provide in vivo evidence that (myo)fibroblasts in wounded skin, lung fibrosis, and intestinal fibrosis may derive from the BM.76 There is evidence that monocytes may represent the major BM-derived cell population that contributes to myofibroblasts in fibrotic lesions, at least in some contexts.77
Circulating fibrocytes express markers of hematopoietic cells (CD34), leukocytes (CD11b, CD13, and CD45), and fibroblast products (collagens I, III, and fibronectin). These cells are distinguished from monocytes/macrophages, dendritic cells, and B cells by their lack of expression of specific markers for these cell lineages. Circulating fibrocytes migrate into injured tissues and have been identified in a number of fibrotic conditions.78 Studies have identified the potential for fibrocytes to participate in wound healing and pathological scarring.78,79 However, the direct contribution of fibrocytes to myofibroblast population during wound healing and fibrosis remains controversial. Evidence for and against fibrocyte-to-myofibroblast differentiation exists.76,80 It is likely that instead of direct differentiation into myofibroblasts, circulating fibrocytes contribute to fibrosis by production of profibrogenic paracrine mediators that target resident cells.
Epithelial–Mesenchymal Transition
EMT refers to as a process through which fully differentiated epithelial cells lose their epithelial characteristics (e.g., apico-basal polarity, polygonal cell shape, and tight and adherens junctions) and acquire properties of mesenchymal cells (e.g., elongated cell shape, increased motility, and contractility). Epithelial cells undergoing EMT are characterized by downregulation of epithelial markers (e.g., E-cadherin and ZO-1) and concomitant upregulation of mesenchymal markers (e.g., FSP1 and α-SMA) in the injured epithelium.81 There is overwhelming evidence that primary epithelial cells cultured in vitro undergo EMT in response to a stimulatory input of soluble growth factors (e.g., TGF-β, EGF, and HGF) and/or ECM components (e.g., collagen).82 However, the concept of in vivo fibrogenic EMT has been challenged by new epithelial lineage tracking studies in a variety of models of kidney, lung, and liver fibrosis in animals.83
 FACTORS REGULATING FIBROBLAST ACTIVATION AND MYOFIBROBLAST DIFFERENTIATION
FACTORS REGULATING FIBROBLAST ACTIVATION AND MYOFIBROBLAST DIFFERENTIATION
Fibroblasts isolated from the site of a healing wound or from fibrotic tissue secrete higher levels of ECM constituents and proliferate more than their normal counterparts isolated from healthy organs.84 Such an increased activity is referred to as “fibroblast activation.” Activated fibroblasts express α-SMA, leading to the term “myofibroblasts.” Fibroblast activation and differentiation into myofibroblasts are regulated by various stimuli, including biochemical factors, biophysical cues from the ECM, and epigenetic modifications.
Growth Factor and Cytokine-Mediated Activation
Fibroblasts become activated by stimulation with cytokines such as TGF-β1, PDGF, and FGF2, which are released from injured epithelial cells, infiltrating mononuclear cells such as monocytes and macrophages as well as the ECM.85–88 TGF-β1 is a pluripotent cytokine that plays a central role in the development of fibrosis. TGF-β1 is sequestered in a latent form (termed latent TGF-β1) in the ECM.89 Latent TGF-β1 becomes activated in a spatially and temporally regulated fashion in response to injury.89 Active TGF-β1 binds to its membrane receptors (TGF-βRI and TGF-βRII) and signals through both Smad-dependent canonical pathway and Smad-independent noncanonical pathway that promote fibrotic gene expression.39 Active TGF-β1 also increases the assembly of stress fibers and FAs that are required for the development of cellular contractility.39 In addition, prototypic cytokines produced by CD4+ T cells such as IL-13/IL-4 and IL-17 exert profibrotic effects on fibroblasts and play a crucial role in the development of fibrosis.90,91
Components of Vascular/Coagulation System
Coagulation proteases such as factor Xa and thrombin activate fibroblasts through receptor-mediated effects elicited by high-affinity thrombin receptor, proteinase-activated receptor (PAR)-1.92 PAR-1 signaling promotes fibroblast proliferation via the autocrine production of PDGF and CTGF, and drives fibroblast differentiation into myofibroblasts via αvβ5-dependent TGF-β activation.93,94 Thrombin upregulates expression of the fibrinolysis inhibitor, plasminogen activator inhibitor (PAI)-1, resulting in increased fibrin matrix.95 Fibrin matrix inhibits surfactant function and contributes to alveolar collapse and traction of remaining airspaces (honeycombing).92 Lysophosphatidic acid (LPA), a platelet-derived molecule during blood coagulation, promotes fibroblast activation and pulmonary fibrosis by activation of latent TGF-β1 through LPA receptor.96 Endothelin-1 (ET-1), an activator of extrinsic coagulation cascade, exerts potent mitogenic and profibrotic effects on fibroblasts.97 Transgenic mice expressing human ET-1 transgene have been shown to develop progressive pulmonary fibrosis.97
Reactive Oxygen Species
Accumulating evidence indicates that oxidative stress and reactive oxygen species (ROS) production, mainly in the form of superoxide and hydrogen peroxide, play a significant role in myofibroblast differentiation.98 The NAD(P)H oxidases of the Nox family have been identified as the enzyme system that is primarily responsible for ROS generation by fibroblasts in response to injury and are recognized as key mediators of myofibroblast differentiation and matrix accumulation in lung fibrosis.99 Nox4, the most abundant Nox isoform in the lung, has been characterized as a “constitutively active” enzyme, meaning that the enzymatic activity of Nox4 is primarily regulated at the level of gene expression. TGF-β1 increases Nox4 expression and ROS production in lung fibroblasts, which mediates TGF-β1–dependent myofibroblast differentiation.99 A similar mechanism for myofibroblast differentiation has been demonstrated in cardiac fibroblasts, renal fibroblasts, and mesangial cells.100,101 Nox4 is also associated with TGF-β1–induced cellular contractility in lung myofibroblasts and cytoskeletal remodeling in vascular smooth muscle cells as well as endothelial cells.99,102,103 Together, these results indicate that Nox-derived ROS regulate fibroblast morphology, contractility, and differentiation.
Mechanical Stress
Fibroblasts respond to mechanical cues, including externally applied forces, interstitial fluid flow, and matrix rigidity sensed through internally generated forces. Externally applied forces such as stretch provoke diverse fibroblast signaling responses, including activation of mitogen-activated protein (MAP) kinases, 104Akt,105 and focal adhesion kinase (FAK). It has been shown that stretch augments TGF-β release and signaling, and promotes the myofibroblast phenotype.106,107 The continuous interactions between cell-generated forces and the resistance of matrix to cellular forces strengthen cell–matrix contacts and develop α-SMA–positive stress fibers characterizing myofibroblast differentiation.39,108 Normal lung fibroblasts grown on polyacrylamide gels with a stiffness grade similar to fibrotic lungs undergo myofibroblast differentiation.109 The effects of matrix stiffening on myofibroblast differentiation may occur through TGF-β–dependent intrinsic and/or TGF-β–independent extrinsic mechanotransduction (see Section “Mechanotransduction in Myofibroblastsinsser”).109,110 Interstitial fluid flow induces fibroblast proliferation, collagen alignment, and fibroblast-to-myofibroblast differentiation in the absence of exogenous mediators.111,112 α1β1 integrin appears to play an important role in the specific response to interstitial fluid flow.111 Taken together, these results suggest that fibroblasts are mechanosensitive and are programmed for matrix production, contraction, and differentiation in the presence of mechanical stimuli.
Epigenetic Regulation
The findings that specific signatures of gene profile in myofibroblasts are “memorized” over passages suggest that epigenetic modifications may be involved in the regulation of myofibroblast differentiation.113 Epigenetic regulation of myofibroblastic phenotype involves DNA methylation, histone modification, and sequence-specific microRNAs (miRNAs). DNA methylation at CpG islands is associated with expression of myofibroblast marker α-SMA in lung cells. Type II alveolar epithelial cells that do not express α-SMA exhibit high levels of methylation at the three CpG islands in the regulatory regions of ACTA2 gene encoding α-SMA protein, whereas lung (myo)fibroblasts exhibit significantly low levels of DNA methylation at these sites.114 Inhibition of DNA methyltransferase (DNMT) induces α-SMA expression, whereas overexpression of DNMT suppresses α-SMA expression in lung fibroblasts.114 Inhibition of DNA methylation activates PPARγ and NF-κB, transcription factors known to suppress ACTA2 gene expression,115 suggesting that modifications of DNA methylation may also indirectly regulate α-SMA expression by inactivation of PPARγ and NF-κB. Histone acetylation has been shown to regulate myofibroblast differentiation. It has been shown that histone deacetylase (HDAC)4 is an essential epigenetic regulator of TGF-β1–induced skin fibroblast-to-myofibroblast differentiation.116 HDAC8 binds to α-SMA and the binding is likely to regulate actin cytoskeleton–derived cellular contractility.117
miRNAs are single-stranded RNA molecules that target multiple mRNAs and induce silencing of multiple transcripts. It has been shown that miR-21 mediates TGF-β1–induced lung myofibroblast differentiation by targeting Smad7, a major inhibitor of TGF-β signaling.118 In contrast, downregulation of miRNA let-7d expression by TGF-β results in increased mesenchymal gene expression (e.g., ACTA2) in multiple epithelial cell lines, indicative of EMT.119 miR-132 inhibits MeCP2 and PPARγ expression, resulting in enhanced α-SMA expression in lung fibroblasts.115
Other Factors
Wnt ligands induce fibroblast activation and collagen synthesis.120 β-catenin, a transcription factor and a downstream signal transducer of Wnt signaling, was found to accumulate in the nuclei of cells located in the fibroblastic foci of IPF lungs.121 Selective inhibition of β-catenin–mediated transcription attenuates lung fibrosis in bleomycin-induced mouse model.122 These studies suggest a crucial role of the Wnt/β-catenin signal in fibroblast activation and lung fibrosis. Integrins have been implicated in the differentiation of proto-myofibroblasts into mature myofibroblasts.123 Integrin α5β1 is associated with α-SMA expression in differentiating myofibroblasts.123 Large clusters of α5β1 are present in the FAs of mature myofibroblasts.123 In addition, environmental stimuli including hypoxia and hyperglycemia as well as direct contacts between fibroblasts and leukocytes have been reported to be associated with fibroblast activation.124,125
 MECHANICAL ASPECTS OF MYOFIBROBLASTS
MECHANICAL ASPECTS OF MYOFIBROBLASTS
Myofibroblast Contractility
Acquisition of contractility similar to smooth muscle cells is a defining feature of myofibroblasts. Myofibroblasts generate intracellular contractile forces by ATP-powered sliding of actin–myosin filaments.126 Compared with Ca2+-regulated rapid and reversible contraction in smooth muscle cells, myofibroblast contraction is relatively slow, sustained, and nonreversible.127 Myofibroblast contractility is primarily regulated by the Rho family of small GTPases (Rho, Rac, and Cdc42) and their downstream targets, primarily myosin light-chain kinase (MLCK) and myosin light-chain phosphatase (MLCP).39 α-SMA expression and incorporation of α-SMA into stress fibers contribute to myofibroblast contractility.128 However, the mechanism by which α-SMA regulates myofibroblast contractile force formation is not clear. Myofibroblast contractility facilitates a normal wound healing process by limiting and closing the exposed surface area of the wound. Myofibroblasts disappear from the wound site when a normal repair process is successfully completed. The persistence of myofibroblasts is associated with aberrant wound repair and leads to tissue fibrosis/scarring. Increasing evidence suggests that myofibroblast contractility may provide a feed-forward mechanism (known as mechanotransduction; see Section “Mechanotransduction in Myofibroblasts”) that sustains fibrosis. Inhibiting myofibroblast contractility by targeting Rho/Rho kinase (ROCK)/actin cytoskeleton signal pathway selectively activates mitochondria-dependent intrinsic apoptotic pathway in myofibroblasts and ameliorates bleomycin-induced mouse lung fibrosis.129
Sensing Matrix Stiffness
The ECM in healthy organs has well-defined mechanical properties that fall within a physiological range.130 Changes in matrix stiffness that occur in pathological states, such as fibrosis and cancer, have profound effects on cell morphology, proliferation, migration, and gene expression.130 Characterization of the mechanical properties of normal and fibrotic lung matrix demonstrates increased matrix rigidity in both human IPF and experimental models of lung fibrosis.131,132 It is believed that protein cross-linking enzymes such as lysyl oxidase (LOXL2) and tissue transglutaminase (TG2) and/or matrix-degrading enzymes such as MMPs regulate the stiffness of ECM, although the precise mechanisms remain to be determined. Fibroblasts sense changes in matrix stiffness by cell–matrix adhesions.133 Transmembrane integrins at FAs act as direct mechanosensors on the cell membrane by providing a physical link between intracellular actin cytoskeleton and the ECM. The cytoplasmic domain of integrins interacts with signaling molecules at the FA sites (e.g., FAK and c-Src). Thus, integrins also act as indirect mechanosensors to regulate cell function and behavior by modulating FA signals.134 Matrix stiffness sensing depends largely on cellular contractility and actin cytoskeleton integrity. Fibroblast contractility generates cytoskeletal tension that serves to transmit mechanical information from the ECM. Disruption of actin cytoskeleton–mediated contractility blocks matrix stiffening–induced myofibroblast differentiation.109
Mechanotransduction in Myofibroblasts
Mechanotransduction is a process in which cells sense mechanical stimuli and convert mechanical signals into biochemical signals. Recent studies suggest that mechanotransduction in myofibroblasts with prolonged survival/resistance to apoptosis may provide a feed-forward mechanism for progression of fibrosis, as in IPF. It has been reported that lung myofibroblast contraction promotes latent TGF-β activation, the most potent fibrogenic cytokine characterized to date, in the extracellular compartment (Fig. 28-3).110,135 In this process, the actomyosin apparatus in myofibroblasts generates contractile forces that are transmitted across the cell membrane to the ECM. The force transmission results in a conformational change of the ECM-bound latent TGF-β1 complex, resulting in the release/exposure of active TGF-β1 from the latent complex. The finding suggests an extrinsic mechanotransduction pathway in which mechanical forces derived from intracellular stress fibers are transduced to the ECM and converted into TGF-β1 fibrogenic signal capable of regulating fibrosis.
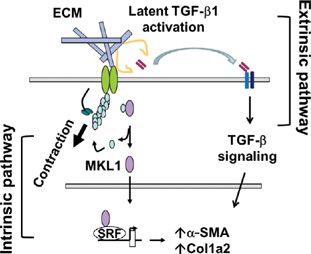
Figure 28-3 Contractile forces promote myofibroblast differentiation via intrinsic and extrinsic mechanotransduction pathways. In the intrinsic mechanotransduction pathway, mechanical stimuli from stiff/fibrotic ECM promote G-actin polymerization into F-actin. This results in the release of MKL1 and its nuclear translocation. MKL1 binds to serum response factor (SRF) in the nucleus to form a transactivation complex and activates fibrotic gene expression that specifies myofibroblast differentiation. In the extrinsic mechanotransduction pathway, actomyosin-generated contractile forces pull against stiff/fibrotic ECM. This results in a conformational change of latent TGF-β1 complex, which releases active TGF-β1 from the latent molecule. Active TGF-β1 then binds to its receptors on the cell membrane and initiates TGF-β signaling that promotes fibrotic gene expression.
In contrast to TGF-β–mediated extrinsic mechanotransduction pathway, an intrinsic mechanotransduction pathway in which the myofibroblast contractile signal is converted into a nuclear signal by transcription factor coactivator megakaryocytic leukemia protein (MKL)1 (also known as MAL/MRTF-A) has also been identified (Fig. 28-3).109
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


