Chapter 53 Exercise-Induced Arrhythmias
Introduction
Exercise-induced arrhythmias are defined as arrhythmias that occur during or shortly after exercise. The observation of this association was first made in 1927 by Bourne, who commented on the development of frequent ventricular premature beats (VPBs) during exercise in patients with suspected coronary artery disease (CAD).1 Later, in 1932, Wilson et al reported the first case of ventricular tachycardia (VT) initiated during exercise.2 Since then, exercise-induced tachyarrhythmias (both supraventricular and ventricular) and bradyarrhythmias have been reported in the literature.3–14
Exercise-induced supraventricular arrhythmias are common. Atrial premature beats have been reported to occur in 5% of “normal” individuals and up to 40% in patients with structural heart disease.15 The incidence rates of atrial fibrillation (AF) and atrial tachycardia (AT) are lower and vary between 0.3% to 1.1% and 0.1% to 2.8%, respectively.7 These have a positive relationship to age and are more frequent in those with underlying structural heart disease.
In addition to conferring a poorer prognosis, the development of ventricular arrhythmias during exercise was formerly considered representative of significant heart disease; as a result, these arrhythmias were more extensively investigated. However, it is now known to not be true for all exercise-induced arrhythmias. The incidence of VPBs during exercise has been shown to vary from 0% to 2.4% in healthy individuals to 31% in patients with structural heart disease.15,16 The association between VPBs during exercise and the presence of ischemic heart disease was recognized early, with up to 50% of patients developing simple VPBs during exercise testing.17,18 In addition, the complexity and number of VPBs were shown to correlate with the degree of associated ST-segment depression, disease severity, and the degree of left ventricular dysfunction, with complex VPBs or repetitive VPBs (salvos, triplets) present in 6% of patients with severe CAD.18,19 In contrast, sustained VT during exercise is relatively uncommon, even among those with underlying CAD (0.15% to 1.7%).20,21 A further study showed the prevalence of complex ventricular premature complexes to be between 15% and 27% in patients with CAD.21a
Although earlier reports clearly identified the association between these arrhythmias and structural heart disease (e.g., a previous myocardial infarction [MI], hypertrophic and dilated cardiomyopathy), more recently, there has been growing interest in inducible ventricular arrhythmias during exercise in those with structurally normal hearts (e.g., long QT syndrome [LQTS] and catecholaminergic polymorphic VT [CPVT]). These arrhythmias occur in seemingly healthy young individuals and can be inherited and associated with sudden cardiac death (SCD). Between 1983 and 1993, Van Camp et al examined 136 deaths among high school and college athletes during or within 1 hour of exercise and found that 100 deaths were likely to have a cardiac etiology: 89% had structural heart disease (hypertrophic cardiomyopathy [56%], anomalous coronary arteries [13%], aortic stenosis [6%], myocarditis [7%], and dilated cardiomyopathy [6%]), but 11% had structurally normal hearts.22 A review of all cases of SCD at the University of Minnesota over a 13-year period found that 5% of SCDs were associated with structurally normal hearts.23 In addition, those with normal hearts were younger (mean age, 35 years), and in 50% the first presentation was SCD following a cardiac arrest.
The clinical presentations of exercise-induced arrhythmias are variable and depend on etiology, the type of arrhythmia, and the presence of structural heart disease. Symptoms range from exertional palpitations, breathlessness, and chest pain to presyncope and syncope. In some, the initial presentation can occur after a cardiac arrest or unexplained SCD. Approximately 6% to 17% of all SCDs occur in association with exercise.24–27
Normal Physiological Effects of Exercise
During exercise, an initial withdrawal of vagal tone occurs, followed by activation of the sympathetic nervous system, which causes an increase in circulating catecholamines.28–31 The effect of this is an increase in heart rate, atrioventricular (AV) conduction, and cardiac contractility, which results in an increase in cardiac output and thus oxygen delivery to organs. These effects are mediated by activation of the α- and β-adrenoreceptors, which directly affect the ion channel currents and electrophysiological properties of cardiac cells. The predominant cardiac receptor is the β1-adrenoreceptor. The intracellular mechanisms of β1-adrenoreceptor activation have been extensively studied and elucidated. β1-adrenoreceptor activation stimulates intracellular G-proteins, which, in turn, activate cyclic adenosine monophosphate (cAMP), protein kinase A, and a number of ion membrane channels. Activation of the pacemaker current (If) and the voltage-dependent calcium (Ca2+) channel (ICa-L and ICa-T) results in an increased rate of phase 4 depolarization of the action potential of pacemaker cells, enhancing normal automaticity and increasing sinus rate. Protein kinase A activation phosphorylates the L-type Ca2+ channel, the ryanodine receptor (RyR) on the sarcoplasmic reticulum, and phospholamban. The net effect of this is increased cytosolic Ca2+ and activation of the sodium-calcium (Na+-Ca2+) exchange pump (Figure 53-1). Increased intracellular Ca2+ causes increased contractility by increasing actin and myosin interaction. Adrenergic stimulation also increases the outward repolarizing potassium (K+) current (Ik) primarily by activation of the slow component of the delayed rectifier current, Iks and the chloride current (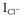 ), resulting in rate-dependent action potential shortening.32 The physiological effects of α-adrenoreceptor activation are less well understood. Activation of the α1-adrenergic receptors reduces K+ conductance primarily from a reduction in the K+ current (Ik) and increases ICa2+ and intracellular Ca2+ via the protein C and inositol phospholipid system.33
), resulting in rate-dependent action potential shortening.32 The physiological effects of α-adrenoreceptor activation are less well understood. Activation of the α1-adrenergic receptors reduces K+ conductance primarily from a reduction in the K+ current (Ik) and increases ICa2+ and intracellular Ca2+ via the protein C and inositol phospholipid system.33
In addition to the direct adrenergic effects associated with exercise, electrolyte and mechanical effects caused by myocardial stretch and baroreceptor activation can affect the electrophysiological properties of cardiac cells; however, the precise mechanism of these is less well understood.34,35
Mechanisms of Arrhythmogenesis During Exercise
In the presence of an appropriate anatomic or electrophysiological substrate, the direct catecholamine effects and indirect effects of exercise can initiate abnormal automaticity, triggered activity, and re-entry.36–38 For example, a ventricular scar caused by a previous myocardial infarct creates a stable anatomic substrate for re-entry. During exercise, catecholamine-sensitive automatic foci can be triggered, causing unidirectional block and re-entry around the scar, resulting in VT. In addition, in the presence of regional wall motion abnormalities, the mechanical stress on the heart during exercise may not be uniform, resulting in abnormal automaticity and heterogeneity of refractoriness, predisposing to re-entry.37 In patients with CAD, exercise-induced ischemia creates an electrophysiological substrate for arrhythmias by its direct effects on ion channel currents and myocyte membrane integrity.39 The resultant alteration in action potential characteristics alters myocyte depolarization, cell-to-cell conduction, and tissue refractoriness, which can cause abnormal automaticity, triggered activity by early afterdepolarizations (EADs) or delayed afterdepolarizations (DADs) and re-entry.39 Genetic mutations of ion channels (LQTS types 1 or 2) or structural proteins involved with intracellular Ca2+ handling (CPVT) create an electrophysiological substrate for exercise-induced ventricular arrhythmias by altering the normal cellular depolarization, repolarization processes, or both during exercise.40,41
Abnormal Automaticity
In diseased states, cells may exhibit automatic activity and may discharge spontaneously in the presence of increased catecholamines during exercise. Abnormal automaticity occurs in patients with myocardial ischemia and electrolyte abnormalities (particularly hypokalemia). Hypokalemia and ischemia reduce the activity of the Na+-K+ adenosine triphosphatase, thereby reducing the background repolarizing current and enhancing phase 4 diastolic depolarization. Abnormal automaticity could be in the form of isolated ectopics or sustained tachycardias. The development of atrial or ventricular ectopy during exercise can trigger other arrhythmias (AF, atrial flutter, AV nodal re-entry tachycardia [AVNRT], AV reciprocating tachycardia, VT, VF) providing a suitable anatomic substrate is present for reentry.3–542
If the automatic focus is sustained, then an AT or VT ensues.42,43 Automatic exercise-induced arrhythmias include atrial or ventricular ectopy, idiopathic outflow tract VT, and arrhythmias associated with myocardial ischemia and hypokalemia.
Triggered Activity
Triggered activity is initiated by afterdepolarizations. DADs occur after repolarization of the action potential and are caused by increased Ca2+ load in the cytosol and the sarcoplasmic reticulum.44 Increased intracellular Ca2+ activates the Na+-Ca2+ exchange pump and the Cl– channel.45,46 The effect of this is to activate the transient inward current (Iti), which causes afterdepolarizations.47 When the inward current is of sufficient threshold, another action potential is initiated, and triggered activity occurs. cAMP has an important role in regulating intracellular Ca2+. Increased catecholamines during exercise activate cAMP and protein kinase A, which increases Ca2+ uptake by the myocyte through the L-type Ca2+ channel and increases release of Ca2+ from the sarcoplasmic reticulum by RyR2 (see Figure 53-1).
Conditions that increase intracellular Ca2+ can initiate DADs and triggered activity. Experimentally, DADs can be initiated during rapid pacing, increased serum Ca2+, digitalis, and endogenous or exogenous catecholamines. This explains the frequent association between exercise (increased catecholamines and heart rate) and the initiation of arrhythmias caused by triggered activity such as outflow tract VTs (e.g., right ventricular outflow tract [RVOT] VT), CPVT, and LQT1 syndrome (torsades de pointes). Conversely, conditions that inhibit the β-receptor–adenyl cyclase cascade, such as vagal stimulation, β-receptor blockade, Ca2+ channel blockade, and adenosine, have been shown to reduce DADs. The effect of adenosine on the ventricles is mediated by the A1-adenosine receptor which causes a conformational change in the Gi-protein, which decreases cAMP levels through its inhibition of adenyl cyclase. The downstream effect of this is reduced protein kinase A–mediated phosphorylation of the L-type Ca2+ channels, RyR2, and phospholamban. The net effect of this is reduced intracellular Ca2+ and thus reduced activity of the Na+-Ca2+ exchange pump and Iti current as well as reduced afterdepolarizations and termination of arrhythmias (see Figure 53-1).47 However, adenosine is unable to terminate ventricular DADs because of cAMP-independent mechanisms such as digitalis or inosine triphosphate–dependent pathways.
“Gain of function” mutations in the sarcoplasmic reticulum Ca2+ release channel (cardiac RyR2) have been identified in patients with the syndrome of CPVT and VF associated with a short Q-T interval.48 Exercise has been shown to potentiate DADs in this group, resulting in polymorphic VT.49
Re-entry
Increases in catecholamines during exercise cause an increase in heart rate and an associated decrease in tissue refractoriness. If an appropriate substrate is present for re-entry arrhythmias (atrial or ventricular scar, accessory pathways, dual AV nodal pathways), the decrease in tissue refractoriness and differential effects of catecholamines on the effective refractory period of different parts of the arrhythmia circuit causes an increase in the excitable gap, which may facilitate the initiation and maintenance of atrial (AVNRT, AV reciprocating tachycardia) or ventricular re-entry arrhythmias.42
Exercise-induced polymorphic VT is thought to be caused by re-entry from nonstationary spiral activity within the ventricular myocardium as a result of the increased heterogeneity of myocardial refractoriness. This can rapidly degenerate into VF. It has been shown that patients with ischemic heart disease and congenital LQT1 have an abnormal adaptation of the Q-T interval during exercise. This would result in the simultaneous presence of areas of depolarized and repolarized myocardium, which creates appropriate conditions for the development of multiple wavelets or nonstationary spiral activity and polymorphic VT.50
Mechanism of Exercise-Induced Bradyarrhythmias
Although the majority of reported cases of exercise-induced arrhythmias are tachycardias, bradycardias have also been reported. Initial case reports described patients with pre-existing conduction disease that gave rise to the development of AV block during exercise.14,51,52 The site of block in these cases was predominantly infra-Hisian. These authors postulated that as the distal conduction system is relatively insensitive to autonomic modulation, during exercise an attenuated increase in AV conduction would occur. Therefore, during exercise, the increased sinus rates would fail to conduct to the ventricles in a 1 : 1 manner.53 Sumoyoshi et al54 reviewed 14 cases of exercise-induced second-degree AV block. The mean atrial rate increased from 62.9 ± 10 beats/min before exercise to 107.1 ± 15 beats/min, but the ventricular rate decreased from 62.9 ± 10 beats/min to 53.4 ± 12.5 beats/min. They reported that the site of block was proximal to the His potential in four patients and intra-His or distal in the remainder. They also noted that AV block was initiated when the atrial rate increased above a critical level governed by the refractory period of the diseased conduction system, which was reproduced with exercise, atrial pacing, and atropine. They therefore concluded that exercise-induced AV block was not purely exercise related but was caused by atrial rate. Other reported mechanisms of exercise-induced AV block are depressed function of the sinoatrial or AV node from hypoxia or ischemia in patients with coexisting lung disease or CAD.11–14 In addition, abrupt cessation of exercise and increased vagal tone could potentially cause bradyarrhythmias.
Conditions Associated with Exercise-Induced Arrhythmias: Structural Heart Disease
Ischemic Heart disease
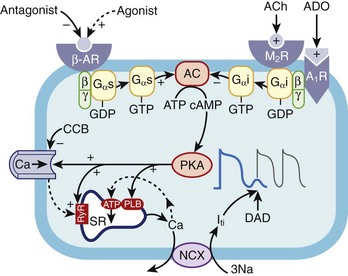
Patients with ischemic heart disease predominantly present with symptoms of chest pain and breathlessness. In some, however, symptoms associated with an arrhythmia frequently provoked by exercise are seen at presentation (Figure 53-2).
Exercise-induced arrhythmias in patients with CAD are multi-factorial. Exercise increases myocardial oxygen demand by increasing cardiac contractility, heart rate, and systolic blood pressure; therefore, if the coronary disease is flow limiting, myocardial ischemia develops. Evidence also suggests that exercise itself increases vasoconstriction at the sites of coronary stenosis. This is thought to be related to endothelial dysfunction and imbalance of endothelium-derived mediators.56,57 Regional differences in myocardial blood flow and oxygen delivery cause changes in regional pH and electrolytes.58,59 Myocardial ischemia results in loss of membrane integrity with K+ efflux, increased Ca2+ influx, decreased amplitude and upstroke velocity of the cardiac action potential, inhomogeneous depolarization of the resting membrane potential, and shortening of the action potential.60 This heterogeneity in the electrophysiological properties (conduction velocity, refractory period) within and around an ischemic zone causes conduction delay, unidirectional block, and re-entry arrhythmias. In experimental studies, following the onset of myocardial ischemia, polymorphic VT and VF was common.61,62 Activation mapping showed principally re-entry arrhythmias.
Exercise itself can promote plaque rupture as the associated increased cardiac contractility and heart rate could create extra stress in the “inflexible” atherosclerotic plaque within the epicardial coronary artery.63 In addition, it has been shown that exercise increases platelet aggregation and could contribute to the development of platelet thrombi over a ruptured plaque.64 Black et al reported clinical, autopsy, and angiography evidence of acute plaque rupture in 13 individuals who had died or had an acute MI during vigorous exercise.63 To determine if a difference in the size of the thrombi was present, Giri et al compared angiography findings in individuals referred for primary angioplasty who had an MI during or within 1 hour of exercise and compared them with patients who had had an MI at rest.65 He showed that a coronary clot of more than 2 mm was present in 64% of the exercise-MI group compared with 35% in the sedentary-MI group and postulated that this difference was caused by differences in the mechanism of plaque rupture. The consequence of acute plaque rupture is myocardial ischemia, which could lead to life-threatening monomorphic or polymorphic VT or VF.66
In patients with a myocardial scar, the mechanism of arrhythmia induction and maintenance is different. The presence of a scar creates an anatomic substrate for re-entry. The resultant VT is monomorphic. These arrhythmias may be initiated during exercise by the sympathetic triggering of automatic ventricular ectopic beats causing unidirectional block and re-entry. Although the majority of arrhythmias associated with exercise-induced ischemia are tachyarrhythmias, bradyarrhythmias caused by myocardial ischemia and CAD have been reported.13
The management of patients with exercise-induced arrhythmias caused by ischemic heart disease depends on etiology. If myocardial ischemia was caused by atherosclerotic coronary disease, then prompt revascularization prevents further arrhythmia recurrence.67,68 If the substrate for the ventricular arrhythmia is a myocardial scar, ablation is a potentially curative treatment if the arrhythmia is incessant and refractory to anti-arrhythmic drugs and implantable cardioverter-defibrillator (ICD) therapy.
Medical management of exercise-induced arrhythmias associated with CAD primarily is with β-blockade. β-Blockers have been shown to be effective in reducing arrhythmia occurrence in patients with ischemic heart disease and after an MI.69 The role of antiarrhythmic drugs in patients with CAD and after an MI was studied in the Cardiac Arrhythmia Suppression Trial (CAST). The hypothesis for this study was that suppression of VPBs with antiarrhythmic drugs would result in a decrease in mortality rate in patients after an MI. They showed that unfortunately, encainide and flecainide increased mortality rates and that other drugs, at best, had only a neutral effect.70 Further meta-analysis in 1993 showed that all antiarrhythmics, with the exception of amiodarone, increased mortality rate in patients with CAD.71,72 Therefore, at present, medical management of exercise-induced arrhythmias associated with ischemic heart disease is with β-blockers or amiodarone.
Arrhythmogenic Right Ventricular Cardiomyopathy
ARVC is characterized morphologically by fibro-fatty infiltration of the myocardium (predominantly of the right ventricle). The most frequent clinical presentation of ARVC is syncope or SCD. ARVC is well recognized as a cause of SCD in the young. A prospective investigation on SCD of the young in the Veneto region in Italy showed that nearly 20% of fatal events in young people and athletes were caused by ARVC. The remainder present with palpitations associated with ventricular ectopy or VT, with a characteristic left bundle branch block (LBBB) morphology. VT associated with ARVC is frequently associated with exercise. Thiene et al found that of the 12 patients identified with ARVC, 10 of them had died suddenly during exercise because of presumed ventricular arrhythmias.73 Rossi et al and Palileo et al initially demonstrated that the re-entry arrhythmia was localized within the abnormal right ventricle.74,75 The presence of widespread disruption of the myocardium by fibro-fatty infiltration creates areas of slow intraventricular conduction and thus represents an ideal substrate for re-entry and ventricular arrhythmias. The precise mechanism for arrhythmogenesis during exercise is not well understood. It is thought that the initiation of the arrhythmia by exercise is related to both hemodynamic and neurohormonal factors. Physical exercise increases right ventricular afterload, which may trigger ventricular arrhythmias by cavity enlargement and increased stretch.76 In addition, progression of disease from the epicardium to the endocardium may cause functional or structural sympathetic denervation, decreased catecholamine reuptake, enhanced sensitivity to catecholamines, and thus increased arrhythmia induction during sympathetic activation during exercise.77 Leclercq et al showed that a stronger sympathetic stimulation was needed to produce sustained VT than to elicit ventricular couplets and nonsustained VT.78
Differentiating idiopathic RVOT VT from VT associated with ARVC is critical because the diagnosis affects long-term prognosis. No pathognomonic feature of ARVC exists; to make the diagnosis, electrocardiography (ECG), echocardiography, right ventricular angiography, cardiac magnetic resonance imaging (MRI), and genetic testing are used. The most common ECG abnormality is T-wave inversion in leads V1 to V3; however, a normal ECG does not exclude ARVC. An ε-wave is found in approximately 50% of those affected and is caused by slowed intraventricular conduction. However, more commonly, signal-averaged ECGs are used to detect late potentials and ε-waves. Although the morphologic features of VT in ARVC and idiopathic RVOT VT are similar, QRS duration may be useful in the differential diagnosis. A QRS duration of 120 ms or more in lead I and a QRS axis less than 30 degrees were found to be predictive of ARVC.79 These differences are caused by the presence of slowly conducting tissue in ARVC and is supported by pacemapping studies in patients with ARVC and idiopathic RVOT VT.79 Transthoracic echocardiography may reveal an enlarged, hypokinetic right ventricle. However, the sensitivity for the detection of ARVC on echocardiography is poor, so cardiac MRI is used frequently. Fatty infiltration and extreme thinning and akinesis of the right ventricular free wall can be seen on cardiac MRI. If the diagnosis is still in doubt after the above investigations, then a transvenous biopsy of the right ventricle can be performed, although it has a low sensitivity in spite of being highly specific for ARVC. A biopsy sample consistent with ARVC would contain more than 3% fat, more than 40% fibrous tissue, and less than 45% myocytes. Exercise stress testing may be helpful in precipitating ventricular arrhythmias in some patients with ARVC; however, response to exercise is variable, and the absence or suppression of PVCs during exercise should not be considered a conclusive factor in terms of its diagnostic exclusion.80 Localized or diffuse wall motion abnormalities in the right ventricle in patients with ARVC may induce ST-segment elevation in response to exercise. Toyofuku and colleagues observed ST-segment elevations of more than 0.1 mV in the right precordial leads and associated right ventricular regional wall motion abnormalities in 65% of patients with ARVC.81 Because exercise-induced ST-segment elevation is a rare phenomenon in normal subjects and in those with idiopathic RVOT VT, this observation may be helpful in diagnosing ARVC.
Left Ventricular Dysfunction
Exercise-induced arrhythmias in patients with left ventricular dysfunction and heart failure are rare and, in fact, exercise has been shown to be beneficial in these patients. A meta-analysis of exercise training in patients with chronic heart failure showed that of the 156 patients in the rehabilitation program, 22 had arrhythmias during exercise.82 One had an episode of AF that spontaneously cardioverted, 11 had premature ventricular beats, and 10 had atrial premature beats. No sustained ventricular arrhythmias were reported. A study from the Lancisi hospital of 154 consecutive patients in a cardiac rehabilitation program found that 21 patients (13.6%) had VPBs, 10 (6.5%) had atrial premature beats, 1 (0.6%) had AF, and 1 (0.6%) had VF requiring DC cardioversion. At the Cleveland Clinic, ECG data were gathered during rest, exercise, and recovery from 2123 patients with left ventricular ejection fraction less than 35%.83 Of these patients, 7% developed exercise-induced ventricular arrhythmias (defined as ventricular triplets, nonsustained or sustained VT, or polymorphic VT or VF) during recovery. This was associated with a 1.5-fold increased risk of death over 3 years, after adjustment for potential confounding factors. These patients are, however, at an increased risk of developing arrhythmias during exercise or recovery. This is thought to be multi-factorial and related to neurohormonal factors (increased sympathetic tone, reduced heart rate variability), electrolyte disturbances (hypokalemia, hypomagnesemia secondary to diuretics), hemodynamic factors (increased end-diastolic pressure, increased afterload, and variations in myocardial stress) and iatrogenic factors (antiarrhythmic drugs).37 Therefore, if arrhythmias develop, it is important to ensure that these patients are on appropriate heart failure treatment (e.g., diuretics, angiotensin-converting enzyme [ACE] inhibitors) and particularly β-blockers, and that serum electrolytes are normal. In addition, patients with sustained ventricular arrhythmias and severe left ventricular systolic dysfunction should be offered ICD therapy.
Mitral Valve Prolapse
Mitral valve prolapse is defined as displacement of mitral valve leaflets by more than 2 mm above the mitral annulus. The reported incidence varies widely between 5% and 38%.84,85 Initial studies suggested that patients with mitral valve prolapse had an increased incidence of ventricular arrhythmias and greater risk of SCD.86,87 A later study by Kilgfield et al demonstrated no increased risk of SCD in patients with mitral valve disease compared with the general population.88 However, patients with mitral valve prolapse do experience palpitations caused by ventricular ectopy and nonsustained VT. In the 1970s, it was appreciated that these ventricular arrhythmias occurred during exercise or during the immediate recovery period.89 In 1986, Butrous et al demonstrated the development of VPBs during the first two stages of the Bruce protocol (increased sympathetic activity), during early recovery after exercise (increased parasympathetic and decreased sympathetic), and during performance of the Valsalva maneuver (rapid increase in parasympathetic activity), and they postulated that the development of VPBs was caused by autonomic imbalance.90
Patients with symptomatic palpitations associated with VPBs frequently respond to β-blockers.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by hypertrophy of the myocardium, particularly the interventricular septum. The overall prevalence of HCM has been estimated to be between 0.05% and 0.2% of the general population. HCM is inherited as an autosomal dominant trait and is attributed to mutations in one of a number of genes that encode for one of the sarcomere proteins. HCM was recognized early as a cause of SCD in young atheletes.91 The myocardium of patients with HCM is characterized by myocardial disarray and myocardial fibrosis, which create an ideal substrate for atrial and ventricular re-entry arrhythmias.92,93 These arrhythmias are frequently initiated during exercise. This is thought to be caused by the development of regional ischemia and hemodynamic effects. The increased muscle mass and narrowed intramural arterioles provoke exercise-induced regional ischemia, which creates heterogeneity in conduction, thus facilitating re-entry, abnormal automaticity, and triggered activity. In addition, during exercise, abrupt increases in outflow tract obstruction increases left ventricular pressure, causing increased stretch, which potentially could induce arrhythmias. McKenna et al investigated the effects of submaximal treadmill exercise on 30 patients with HCM.94 One patient (3%) had no arrhythmia, 14 (46%) had supraventricular tachycardia or paroxysmal AF, 13 (43%) had multi-form or paired ventricular extrasystoles, and 8 (26%) had VT. Routine echocardiographic or hemodynamic measurements did not predict the development of serious ventricular arrhythmias. It was concluded that asymptomatic exercise-induced ventricular arrhythmia is a common occurrence in patients with HCM.
Conditions Associated with Exercise-Induced Arrhythmias Without Structural Heart Disease
Approximately 10% of patients presenting at the hospital with VT have no obvious evidence of underlying structural heart disease or CAD.95
Catecholaminergic Polymorphic Ventricular Tachycardia
Familial CPVT is a rare cause of exercise-induced VT. Sporadic cases were initially reported in the 1970s; however, in the past decade, a genetic etiology has become apparent.96 The prevalence of CPVT is unknown but is estimated to be approximately 1 per 10,000. CPVT diagnosis is frequently missed because individuals are often young with structurally normal hearts. Approximately 30% of probands present with a family history of stress-related syncope, seizure, or SCD.97 SCD can be the first manifestation of CPVT.
CPVT is inherited as an autosomal dominant or recessive trait, usually with high penetrance. In the dominant form, the disease-causing gene has been mapped to chromosome 1q42 to 43, which codes for the cardiac RyR gene RyR2, which is a tetrameric intracellular Ca2+ release channel on the sarcoplasmic reticulum required for excitation-contraction coupling. In addition, RyR2 gene mutations have been identified in patients affected by ARVC2, a variant form of ARVC.98 Mutations of the RyR2 channel result in a “gain of function” thought to be caused by either the decreased binding affinity of the stabilizing FKBP 12.6 protein or the increased sensitivity of the channel to Ca2+ and increased propensity to spontaneous release of Ca2+ from the sarcoplasmic reticulum.99–102 In the recessive form, a missense mutation on the locus of chromosome 1p13-21 has been identified.102,103 This codes for CASQ2, which is the major Ca2+ reservoir within the sarcoplasmic reticulum of cardiac myocytes. Abnormalities in the RyR2 channel proteins and CASQ2 result in increased intracellular Ca2+ and cytosolic Ca2+ overload; this, in turn, generates a net transient inward current (Iti), which underlies diastolic membrane depolarization, which, if sufficient, could reach the threshold for Na+ current activation and triggered activity.44
During exercise, increased catecholamines activate the β1-receptors, cAMP, and protein kinase A, which triggers Ca2+ release from the sarcoplasmic reticulum. In patients with CPVT, the abnormal RyR2 channel and CASQ2 result in an abnormally high intracellular Ca2+ release, causing DADs or triggered activity and polymorphic VT. Liu et al, in their transgenic CPVT murine model, demonstrated the enhancement of DADs after β-adrenergic stimulation and the development of multiple triggered action potentials.104 Subsequently, a number of other studies have confirmed these findings.105–108 In humans, Paavola et al showed that in humans, those with the RyR2 mutation developed more DADs after the administration of adrenaline compared with control subjects.49
The clinical manifestations of CPVT are exercise-induced polymorphic, monomorphic, or bidirectional VT in patients with structurally normal hearts. Although, classically, CPVT is associated with bi-directional VT, this is the presenting arrhythmia in only 35% of patients.109 The ventricular arrhythmias that occur during exercise stress testing appear quite consistently at heart rates of 110 to 130 beats/min. They initially start as polymorphic premature ventricular complexes, and then at increasing workload, an increased complexity of ventricular arrhythmia is observed (i.e., bigeminy and couplets leading to bi-directional VT, polymorphic VT, or both). When the exercise stops, the arrhythmias gradually disappear. Monteforte et al analyzed ventricular arrhythmias developing during exercise stress testing in 61 consecutive patients with CPVT before therapy.110 They demonstrated a positive direct correlation between the coupling interval of ventricular arrhythmias and the preceding R-R interval, supporting the view that triggered activity is the mechanism underlying arrhythmias in CPVT.
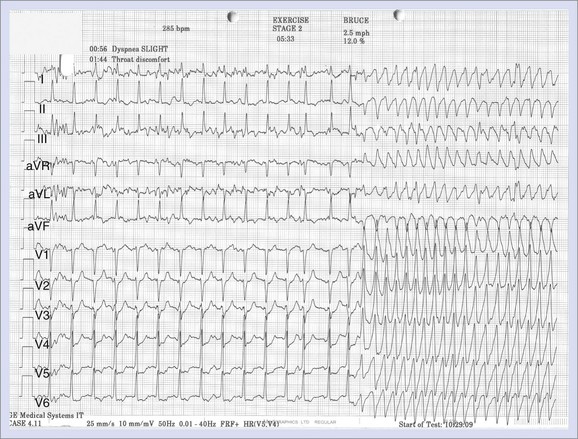
The mechanism for bi-directional VT has been the subject of much debate. Since its initial description in 1922, several hypotheses have been suggested, including two separate foci and re-entry.111,112 Cerrone et al demonstrated, in their CPVT mouse model, the development of monomorphic, polymorphic, and bi-directional VTs during adrenergic stimulation and Ca2+ overload.108 Endocardial mapping during the arrhythmia showed that the foci were predominantly from the specialized conduction system. Monomorphic VT was unifocal, and polymorphic VT was multifocal, later degenerating into re-entry and VF. A case report by Mok et al demonstrated the development of unifocal ventricular ectopy, followed by polymorphic VT and then bi-directional VT during adrenaline infusion (Figure 53-3).113
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


