Fig. 16.1
Atherosclerotic lesions with luminal thrombi. Coronary plaque features responsible for acute thrombosis comprise three different morphologies: rupture, erosion, and calcified nodules. Ruptured plaques are thin fibrous cap atheromas with luminal thrombi (Th). These lesions usually have an extensive necrotic core (NC) containing large numbers of cholesterol crystals and a thin fibrous cap (<65 μm) infiltrated by foamy macrophages and T-lymphocytes. The fibrous cap is thinnest at the site of rupture and consists of a few collagen bundles and rare smooth muscle cells. The luminal thrombus is in communication with the lipid-rich necrotic core. Erosions occur over lesions rich in smooth muscle cells and proteoglycans. Luminal thrombi overly areas lacking surface endothelium. The deep intima of the eroded plaque often shows extracellular lipid pools, but necrotic cores are uncommon; when present, the necrotic core does not communicate with the luminal thrombus. Inflammatory infiltrate is usually absent, but if present, is sparse and consists of macrophages and lymphocytes. Calcified nodules are plaques with luminal thrombi showing calcific nodules protruding into the lumen through a disrupted thin fibrous cap. There is absence of an endothelium at the site of the thrombus, and inflammatory cells (macrophages and T-lymphocytes) are absent (From Virmani et al. [2]. Reprinted with permission from Wolters Kluwer Health)
Plaque ruptures develop in a lesion with a large necrotic core and an overlying thin disrupted fibrous cap, which is heavily infiltrated by macrophages and T-lymphocytes; a luminal thrombus develops because of physical contact between the flowing blood with the thrombogenic necrotic core. In contrast, erosions are characterized by a luminal thrombus superimposed on a proteoglycan-rich plaque containing mostly smooth muscle cells (SMCs) with few inflammatory cells [4, 5]. Calcified nodule, the least frequent cause of coronary thrombi, typically contains calcified plates along with calcified nodules that penetrate the lumen, which leads to the disruption of the collagen and endothelial layers [2].
Plaque Rupture and Its Precursor Lesion – The Thin Cap Fibroatheroma (TCFA)
Thin-cap fibroatheroma (TCFA), traditionally designated as vulnerable plaque, is recognized by its morphologic characteristics that resembles ruptured plaque, and the main difference is the absence of a luminal thrombus and an intact thin fibrous cap [2]. TCFAs generally contain a relatively large necrotic core, although smaller than seen in ruptured plaques; an overlying thin fibrous caps with varying degrees of macrophages and lymphocytic infiltration and few to absent SMCs (Fig. 16.2). The fibrous cap thickness as an indicator of plaque vulnerability is defined to be ≤65 μm since the thinnest portion of the remnant ruptured cap was 23 ± 19 μm, with 95 % of the TCFA caps measuring ≤65 μm [3]. Although there are similarities, TCFAs differ from ruptured plaques on the basis of trends toward a smaller necrotic core, fewer macrophages within the fibrous cap and less calcification (Table 16.1) [5].
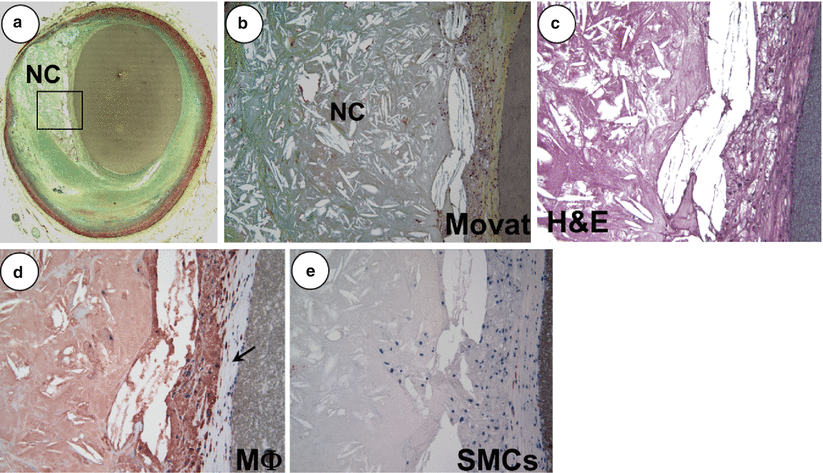
Fig. 16.2
A non-haemodynamically limiting thin-cap fibroatheroma. (a) Low power view of a thin-cap fibroatheroma (TCFA) with a relatively large eccentric necrotic core (NC) and an overlying thin fibrous cap (<65 μm); the greyish material in the lumen is barium gelatin (Movat pentachrome, ×20). (b) Image of the boxed area in panel (a) showing an advanced necrotic core with a loss of matrix containing numerous cholesterol clefts and cellular debris (×100). (c) High power view of a fibrous cap heavily infiltrated by inflammatory cells (hematoxylin and eosin, ×200). (d) Numerous CD-68 positive macrophages are localized to the fibrous cap (arrow) (x200). (e) The fibrous cap shows rare a-actin positive smooth muscle cells (SMCs) (×200) (From Kolodgie et al. [6]. Reprinted with permission from BMJ Publishing Group Ltd)
Table 16.1
Morphologic characteristics of plaque rupture and thin-cap fibroatheroma
Plaque type | Necrotic core (%) | Fibrous cap thickness (μm) | Mϕs (%) | SMCs (%) | T-lymph | Calcification score |
|---|---|---|---|---|---|---|
Rupture | 34 ± 17 | 23 ± 19 | 26 ± 20 | 0.002 ± 0.004 | 4.9 ± 4.3 | 1.53 ± 1.03 |
TCFA | 23 ± 17 | < 65 μm | 14 ± 10 | 6.6 ± 10.4 | 6.6 ± 10.4 | 0.97 ± 1.10 |
p value | ns | 0.005 | ns | ns | 0.014 |
It is generally a prevailing thought that rupture of fibrous caps occurs at their weakest point, often near shoulder regions. However, this is not always the case in our experience as we have observed an equivalent number of ruptures occurring at the mid portion of the fibrous cap. These differences were even greater when death occurred during exertion or at rest; the former ruptured in the mid fibrous cap and the latter in the shoulder region [8]. Therefore, it is reasonable to speculate that different processes may be involved as catalysts for the rupture event. It has been shown that macrophages in the fibrous cap release matrix metalloproteinase (MMP) [9] which further weaken the cap, along with high shear stress [10], macrophage cell death [11], and microcalcification and iron accumulation within the fibrous cap [12], all contribute to the eventual disruption of the cap.
Once the fibrous cap ruptures and the necrotic core contents are exposed to the flowing blood in the lumen, coagulation cascade is activated in response to the exposure of lipids and tissue factors (TF) which are present in the core. The luminal thrombus is rich in platelet (white thrombus) near the actual rupture site, while at sites proximal and distal to the rupture, there is propagation of the thrombus which is rich in erythrocytes that alternate with regions of fibrin and platelets (red thrombus). A study of aspirated thrombi from acute myocardial infarction patients examined by scanning electron microscopy (SEM), confirmed a decrease in platelet content and an increase of fibrin and red cell content in accordance with ischemic time duration [13].
Comparison of Morphologic Features Between Plaque Rupture, TCFA, Erosion and Stable Plaques
We have quantified several morphologic parameters in various human coronary plaque types, including culprit lesions (Table 16.2). The number of cholesterol clefts in the necrotic core, vasa vasorum, and macrophages are significantly greater in ruptured plaques than erosions or stable plaque with >75 % cross-sectional luminal narrowing. There was a greater accumulation of hemosiderin in ruptures as compared to TCFAs.
Table 16.2
Comparison of necrotic core size, number of cholesterol clefts, macrophage infiltration, number of vasa vasorum, and hemosiderin laden macrophages in culprit plaques
Plaque type | Necrotic core (%) | Cholesterol clefts (%) | Mϕs in fibrous cap (%) | Mean number of vasa vasorum | Mean number of hemosiderin laden Mϕs |
|---|---|---|---|---|---|
Rupture | 34 ± 17a | 12 ± 12b | 26 ± 20c | 44 ± 22d | 18.9 ± 11e |
TCFA | 24 ± 17 | 8 ± 9 | 14 ± 10 | 26 ± 23 | 4.4 ± 3.6 |
Erosion | 14 ± 14 | 2 ± 5 | 10 ± 12 | 28 ± 18 | 4.3 ± 4.7 |
Stable | 12 ± 25 | 4 ± 6 | 3 ± 0.7 | 13 ± 9 | 5.0 ± 9.3 |
p value | a0.003 vs. erosion | b0.002 vs. erosion | c0.005 vs. TCFA | d0.07 vs. TCFA | e0.001 vs. TCFA |
a0.01 vs. stable | b0.04 vs. stable | c<0.0001 vs. erosion | d0.02 vs. erosion | e<0.0001 vs. erosion | |
c0.0001 vs. stable | d0.01 vs. stable | e0.03 vs. stable |
The extent of luminal narrowing varies with underlying plaque morphology. TCFAs and fibroatheromas have the least luminal narrowing while lesions with acute plaque rupture, hemorrhage, or healed plaque rupture sites show the most stenosis [6]. The vast majority of TCFAs (approximately 80 %) have <75 % cross sectional area luminal narrowing (equivalent to <50 % diameter stenosis) (Fig. 16.3a). Forty-six percent of healed plaque rupture and 43 % of acute rupture show the severe luminal narrowing (i.e., >75 % cross-sectional area narrowing); in contrast, only 26 % of TCFAs show severe luminal narrowing.
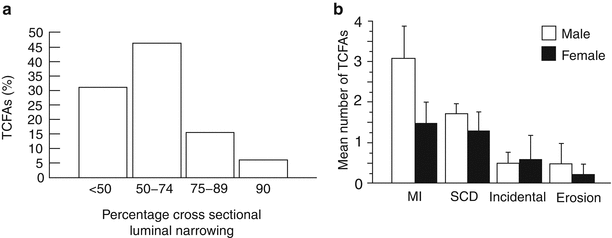
Fig. 16.3
The percentage off thin-cap fibroatheromas (TCFAs) stratified by the degree of luminal narrowing (a) and number of TCFAs in various coronary syndromes (b). (a) Approximately 80 % of these lesions have <75 % cross sectional area luminal narrowing. (b) TCFAs are most frequent in patients dying with acute myocardial infarction (MI), followed by sudden coronary death (SCD) victims and incidental disease, but are uncommon in patients dying from erosion ((a) From Kolodgie et al. [6]. Reprinted with permission from BMJ Publishing Group Ltd. (b) From Burke et al. [14]. Reprinted with permission from Elsevier Limited)
The mean necrotic core size in lesions from sudden coronary death victims is independent of luminal narrowing and is greatest in plaque ruptures, followed by TCFAs, erosion and fibroatheromas (Table 16.2). In a detailed morphometric analysis of ruptured plaques, 80 % of necrotic cores were larger than 1.0 mm2 and comprised >10 % of plaque area in approximately 90 % of the lesions. In 65 % of plaque ruptures, the necrotic core occupied >25 % of plaque area; in contrast, 75 % of TCFA had necrotic cores >10 % of plaque area. The mean cross-sectional luminal narrowing in TCFAs was 71 %, with necrotic core representing 10–25 % of the lesion. On the other hand, the length of the necrotic core in ruptures and TCFAs was similar, varying from 2 to 22.5 mm, with a mean of 8 and 9 mm, respectively [15]. In terms of the circumferential involvement of the necrotic core, 75 % of TCFAs show >120° of the intima involved by a necrotic core [6].
Incidence of TCFA in Various Coronary Syndromes and Its Distribution in the Coronary Arteries
When we analyze the number of TCFAs by plaque morphology, the highest incidence is observed in plaque rupture followed by healed rupture and stable plaques; the incidence being the least in erosion [7]. In patients presenting with acute myocardial infarction, the incidence of TCFAs is highest in males with a mean of 3 per heart, with half as many in women (Fig. 16.3b) [14]. In patients dying suddenly, although the incidence of TCFAs was greater in men than in women, the difference was not statistically significant. In those dying from incidental death or plaque erosion, the incidence of TCFAs was lower as compared to those dying from acute myocardial infarction or sudden coronary death, without any differences observed between the two sexes.
TCFAs and plaque ruptures show a predilection for certain anatomic locations within the coronary tree. The majority of acute and healed ruptures and TCFAs are concentrated in the proximal and mid portion of the three major coronary arteries, and are rarely observed in the distal portion of these arteries (Fig. 16.4) [7]. By far, the most frequent location is the proximal portion of the LAD coronary artery, with sites in the proximal left circumflex and mid to proximal right coronary arteries being half as common.
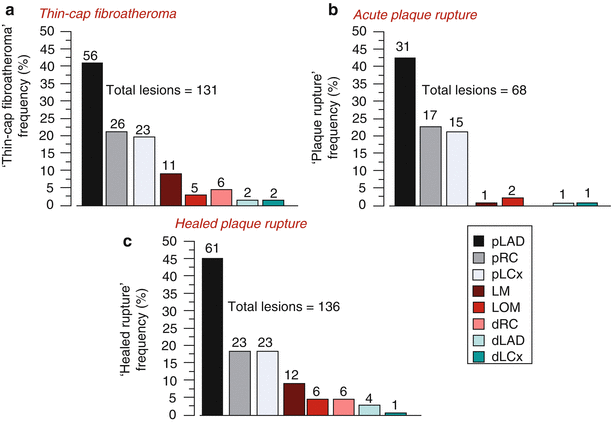
Fig. 16.4
Frequency and location of unstable lesions: thin-cap fibroatheromas, acute and healed ruptures in the coronary circulation. Bar graphs illustrating the frequency and location of thin-cap fibroatheromas (a), plaque rupture (b) and healed rupture (c) in the human coronary circulation. The proximal left anterior descending coronary artery (pLAD) represents the first 5.0 ± 2.5 cm of the artery measured to the second diagonal branch. The proximal right coronary (pRC) includes the distance to the right marginal branch of the right coronary artery (first 5 cm). The proximal left circumflex (pLCx) is the level to the first left obtuse marginal (LOM) origin and is the most variable in length. LM left main, dRC distal right coronary, dLAD distal left anterior descending, and dLCx distal left circumflex artery. Note, the majority of thin-cap fibroatheromas, ruptures, and healed rupture occur in the pLAD (From Kolodgie et al. [7]. Reprinted with permission from Wolters Kluwer Health)
It has been observed that the fibrous cap thickness is dependent on the extent of macrophage infiltrate; the thinner the fibrous cap, the greater the number of macrophages, whereas the thicker the fibrous cap, the fewer the macrophages. In serial sections of TCFAs, the necrotic core was located deep within the plaque; i.e., away from the lumen but over a relatively short distance of <1.4 mm, the lipid core approaches the lumen. Therefore, histologically the presence of suspected TCFAs may be better identified through serial sectioning [16].
Intraplaque Hemorrhage and Expansion of the Necrotic Core
Intraplaque hemorrhage in non-culprit sites of the coronary vasculature is more common in hearts with ruptures versus stable plaques without luminal thrombi. The mean ± SD number of acute hemorrhages in non-culprit lesions from patients with rupture was 2.5 ± 1.3 versus none in erosion (p = 0.0001) and 0.05 ± 0.6 in stable plaques (p = 0.04) [2]. Evidence of prior hemorrhages as analyzed by anti-glycophorin A staining (a protein specific to erythrocyte membranes) was significantly greater in TCFAs than in fibroatheromas with early or late necrotic cores or lesions with pathologic intimal thickening. The degree of glycophorin A staining correlated with both necrotic core size and the extent of macrophage infiltration suggesting that erythrocytes may directly contribute to necrotic core expansion and lesion instability [17].
Ira Tabas has reported in the mouse model that it is the apoptotic cell death of foamy macrophages, which contribute to the formation of necrotic core that contains phospholipids, cholesterol esters and free cholesterol [18]. Also, impaired phagocytic efficiency of macrophages to effectively clean up apoptotic bodies i.e., defective efferocytosis further contributes to the progression of necrotic core [17, 18]. In man, we have observed that foamy macrophages infiltration into lipid pools and apoptotic cell death of these cells leads to early necrotic core formation. However, it is not only apoptotic foamy macrophages that contribute to the accumulation of free cholesterol but other sources should also be considered that may expand the necrotic core. Studies from our laboratory have shown that hemorrhage into a plaque is a common event that is most frequently observed in cases of plaque rupture and severe coronary artery disease. The membranes of red blood cells are rich in lipid content and constitute 40 % by weight that exceeds all other cell types [19]. Excess membrane cholesterol of red blood cells can phase separate and form immiscible membrane domains consisting of free cholesterol arranged in a tail-to tail orientation favoring crystal formation [20]. The insudation of lipoproteins within the plaque matrix, dead and dying foamy macrophages, along with accumulation of erythrocytes membranes that are rich in free cholesterol all collectively influence both the biochemical composition and size of the necrotic core. The origin of intraplaque hemorrhage is also debated between those who believe that it is the influx of blood from the luminal through plaque fissuring to others who propose that it is leakage from intraplaque vasa vasorum. However, we favor of the latter since it is often the case that intraplaque erythrocyte extravasation is seen in the absence of plaque fissure and hemorrhage is associated with a high density of vasa vasorum within and close to the necrotic core (Fig. 16.5). Further, some reports demonstrated dysmorphogenesis of the vasa vasorum with absence of mural cells and presence of poor endothelial cell junctions [22]. It is possible that these immature or leaky vessels also allow diffusion of plasma molecules and diapedesis of leucocytes as well as erythrocytes [23], which may serve as the driving force of further centripetal angiogenesis from the adventitia [24].
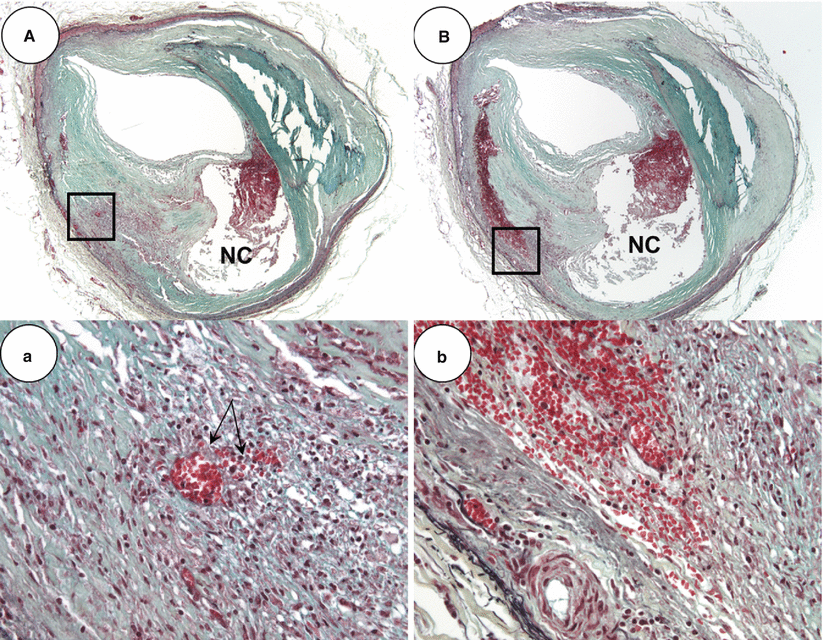
Fig. 16.5
Thin-cap fibroatheroma with recent intraplaque hemorrhage. (A) Low (×20) and higher power (×200) views, respectively, of a thin-cap fibroatheroma with recent hemorrhage. Note, in (a) there is spillage of erythrocytes from surrounding intraplaque vasa vasorum (arrows). (B) Serial section of the lesion in (A) shows a large pool of extravasated erythrocytes with proximate micro-vessels. Panel (b) is a high power view of boxed area in (B) (From Virmani et al. [21]. Reprinted with permission from Wolters Kluwer Health)
Hemoglobin Toxicity and Oxidative Stress
Intraplaque hemorrhage could potentially provoke recruitment of inflammatory cells [25]. The precise signaling pathways for the cell migration are not fully understood but it is postulated that proteins in coagulated blood could contribute to inflammatory cell activation [26]. However more importantly, recent studies have elucidated the significance of oxidative stress associated with extravasated erythrocytes providing a rapid influx of hemoglobin (Hb) and continued inflammation in the intraplaque area. Free Hb can also bind and inactivate nitric oxide (NO), a potent signal molecule that plays a critical role in the relaxation of smooth muscle and reduced expression of endothelial adhesion molecules, eventually leading to cell injury [27].
A major protective mechanism against Hb toxicity is mediated by haptoglobin (Hp), an abundant serum protein whose major function is to bind excess Hb. In atherosclerotic plaques, the primary route for clearance of the Hb-Hp complex involves the CD163 receptor expressed on immunosuppressive macrophages with M2 polarized phenotype [28]. Another method for protection is provided by the presence of heme oxygenase-1 (HO-1) which has anti-oxidative, anti-inflammatory, anti-apoptotic and possibly immune-modulatory properties and is expressed in endothelial cells, macrophages and SMCs [29]. In ApoE deficient mouse models with over expression of heme oxygenase-1, decreased atherosclerotic lesions have been reported [30].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


