an oxygen debt as is seen with skeletal muscle. In a steady state, cardiac metabolic activity is thus accurately measured by myocardial oxygen demand (MVO2). The total metabolism of an arrested, quiescent heart is approximately 1.5 mL/minute per 100 gm, as required to support the physiologic processes not directly associated with contraction. In contrast, a beating canine heart has MVO2 ranging from 8 to 15 mL/minute per 100 gm.1, 2, 3
Table 24.1 Myocardial Oxygen Consumption Components Total: 6-8 mL/min per 100 gm | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
during tachycardia because of simultaneously increasing demand (tachycardia) and compromised flow for the subepicardium.3
Table 24.2 Determinants of MVO2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
substances, and neurohumoral stimuli. Large-conduit arteries are unaffected by myocardial metabolites because of their extramural location, but can produce episodic increases in resistance during severe focal or diffuse contraction (vasospasm) in the absence of atherosclerosis. One exception is myocardial bridging, in which intramyocardial vessel segments may offer increased resistance during systole owing to mechanical compression of the bridged segment during ventricular contraction.
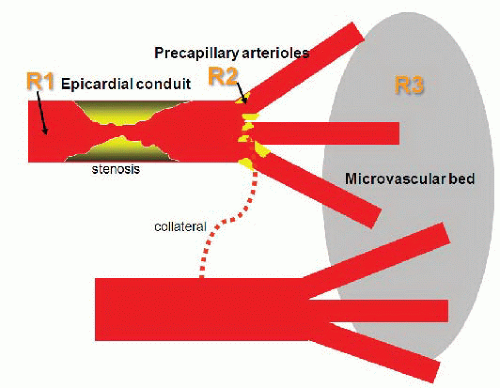 Figure 24.1 Diagram of coronary resistances. The epicardial arteries (R1) normally have negligible resistance until an atherosclerotic narrowing occurs (top artery). The precapillary arterioles (R2) regulate most of the coronary flow to the microvascular bed (R3). Diseased epicardial vessels are often connected to normal blood flow regions by collateral channels. (Modified from Dr. Bernard De Bruyne with permission.) |
stimuli include the reactive hyperemia that follows transient coronary occlusion, exercise, and the administration of various pharmacologic agents. Coronary flow reserve is expressed as the ratio of maximal hyperemic flow to resting coronary flow—a ratio that averages from 4 to 7 in experimental animals and from 2 to 5 in man.11,12 In experimental animal studies, increasing conduit stenosis (R1) produces a predictable decline in coronary flow reserve, beginning at about a 60% artery diameter narrowing. At diameter stenoses of >80% to 90%, all available coronary reserve has been exhausted and resting flow begins to decline13, 14, 15 (Figure 24.2). (Factors responsible for reduced CVR in the absence of epicardial stenosis are listed in Table 24.5.) This relationship between increasing stenosis severity and reduced available flow reserve has been used in assessing the effective physiologic severity of any given coronary lesion and forms the basis of many noninvasive test modalities for ischemia. In clinical practice, however, for an individual patient, this relationship is unpredictable given the complex three-dimensional anatomy, imprecise correlation between angiographic estimate of diameter reduction owing to stenosis and true lumen cross-sectional area, and unknown status of microcirculation.
Table 24.3 Regulation of Coronary Circulation | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Table 24.4 Mediators of Coronary Vasodilation | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
obstruction that may be superimposed by platelet aggregation and thrombosis compromising lumen area, a process active in acute coronary syndromes (ACSs).16
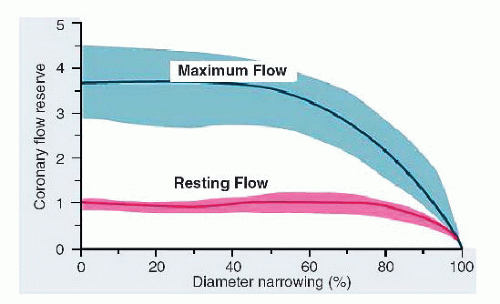 Figure 24.2 Coronary flow reserve expressed as ratio of maximum to resting flow plotted as a function of percent diameter narrowing. With progressive narrowing, resting flow does not change (dashed line), whereas maximum potential increase in flow and coronary flow reserve begin to be impaired at approximately 50% diameter narrowing. The shaded areas represent the limits of variability of data about the mean. (From Gould KL, Lipscomb K, Hamilton GW. Physiologic basis for assessing critical coronary stenosis: instantaneous flow response and regional distribution during coronary hyperemia as measures of coronary flow reserve. Am J Cardiol 1974;33:87-94.) |
Table 24.5 Factors Responsible for Microvascular Disease and Reduction of Coronary Flow Reserve | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
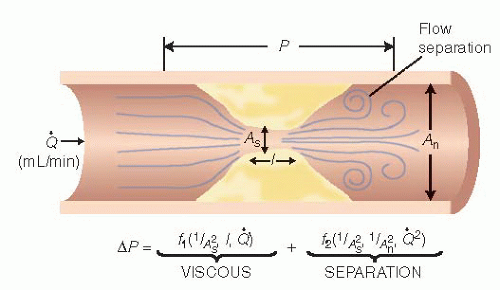 Figure 24.3 Diagrammatic illustration of the Bernoulli equation. ΔP, pressure gradient; As, area of the stenosis; An, area of the normal segment; L, stenosis length; Q, flow; f1, viscous friction factor (f); f2, separation coefficient (s). See text for details. |

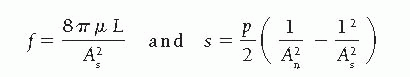
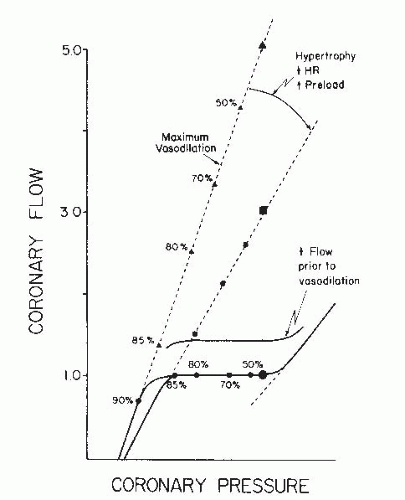 Figure 24.4 Resting and maximally vasodilated coronary pressure-flow relationships. Coronary flow reserve, the ratio of maximally vasodilated flow to resting flow, is a complex function of the actual position of the maximally vasodilated and resting flow curves. The slope of the maximal vasodilation curve can be shifted by hypertrophy and changes in hemodynamics as can the basal flow be altered by similar events, thus explaining different CFR (maximal vasodilation flow/basal flow ratio) under different conditions and in different patients. (With permission from Klocke FJ. Measurements of coronary flow reserve: defining pathophysiology versus making decisions about patient care. Circulation 1987;76:1183.) |
Table 24.6 Thrombolysis in Myocardial Infarction Flow Grade | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
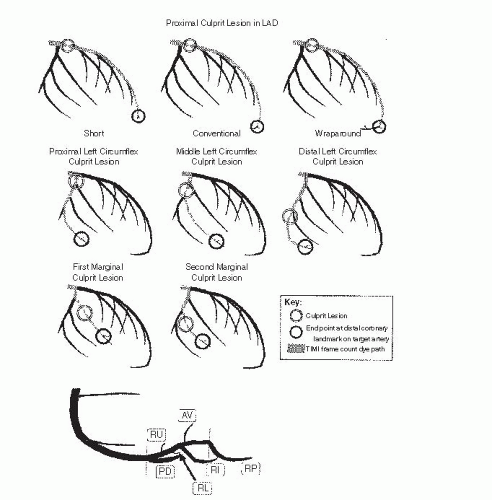 Figure 24.5 Top. Anatomic landmarks used for TIMI frame counting in the left anterior descending coronary artery. The distalmost branch in the left anterior descending coronary artery (referred to as the pitchfork, mustache, or whale’s tail) usually occurs at the apex of the heart. In a wraparound left anterior descending coronary artery, the branch closest to the apex of the heart is used. Second and third rows. Anatomic landmarks used for TIMI frame counting in the left circumflex coronary artery. The artery used for TIMI frame counting is the artery with the longest total distance along which dye travels in the left circumflex coronary artery system and yet passes through the culprit lesion. When the culprit lesion is proximal to two arteries with equal total dye-path distances, the artery that arises more distally from the left circumflex coronary artery is used. For example, when the culprit lesion is located in the proximal left circumflex coronary artery, the marginal branch with the longest total dye-path distance is used, regardless of whether it is the first, second, or third marginal branch. If these second and third marginals have equal total dye-path distances, the third marginal branch is the target artery. The target artery is always the first marginal branch when the culprit lesion is in the first marginal and, likewise, always the second marginal branch when the culprit lesion is in the second marginal. In left and balanced dominant systems, the target artery is no farther distal than the marginal branch that lies at the border of the inferior and lateral walls, usually the third or fourth marginal. The anatomic endpoint is the distalmost branch in the target artery. Usually, this endpoint branch can be found at approximately the midpoint of the distal third of the artery (five-sixths of the distance down the vessel from its origin), but occasionally is located just before the termination of the artery. Bottom. Anatomic landmarks used for TIMI frame counting in the RCA. The distal landmark is the first branch arising from the posterior lateral extension of the RCA after the origin of the posterior descending artery, regardless of the size of this branch. As shown, this branch will often be located just distal to the bifurcation and may be oriented either superiorly (RU) or inferiorly (RL). In some cases, this branch will lie farther along the extension of the distal RCA and either will course superiorly as the AV nodal artery (AV) or will be oriented inferiorly as the right inferior branch (RI). In the event that a very proximal posterior descending stenosis is the culprit lesion, the first branch off the posterior descending artery after the stenosis is the endpoint. Infrequently, the distal portion of the posterior descending artery is supplied by a proximally arising acute marginal branch, and the proximal portion of the posterior descending artery arises at the base of the heart. In these cases, it is the extension of the distal RCA past the posterior descending artery at the base of the heart, and not the proximal acute marginal branch, that is used for determining the TIMI frame count. In patients with left-dominant anatomy, the TIMI frame count endpoint is the distalmost branch of the RCA once it is no longer in the atrioventricular groove. (Adapted with permission from Gibson CM, Cannon CP, Daley WL, et al. TIMI frame count: a quantitative method of assessing coronary artery flow. Circulation 1996;93:879-888.) |
Table 24.7 Reference Values for Thrombolysis in Myocardial Infarction Frame Counts | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
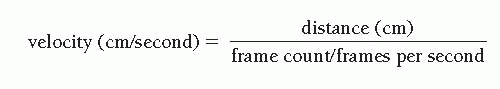
grading to define successful angiographic reperfusion following primary angioplasty for acute myocardial infarction.29
Severe transient bradycardia after IC adenosine occurs in <2.0% of patients, coronary spasm during passage of the Doppler guide wire in 1%, and ventricular fibrillation during the procedure in 0.2% of patients.36
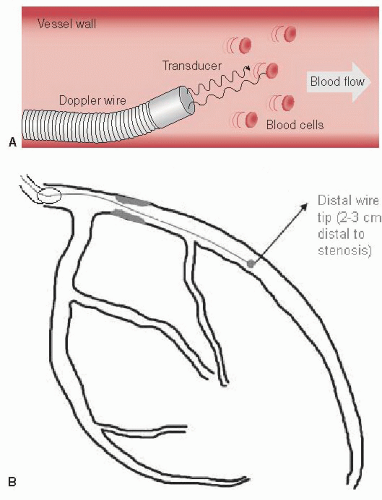 Figure 24.6 A. Schematic depiction of a Doppler flow wire emitting ultrasound signals along the direction of the vessel with return of the signals after interacting with flowing red blood cells to produce a Doppler-derived blood flow velocity in a coronary artery. B. Depiction of a coronary tree with a stenosis and Doppler flow wire advanced through the guiding catheter, across the stenosis and with the tip distal to the stenosis. In this way, coronary flow reserve (CFR) can be determined to evaluate the significance of the stenosis. The same setup can be used to measure fractional flow reserve (FFR) if a pressure wire is used instead of a Doppler flow wire.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|