balloon. While this design allowed for relatively straightforward deployment, the rigidity of this initial stent design made it difficult to deliver this device to the coronary vasculature. In 1989 a design modification was made by Richard Schatz, consisting of the placement of a 1 mm central articulating bridge connecting the two rigid 7 mm slotted segments,5 creating the 15 mm Palmaz-Schatz stent (Johnson and Johnson Interventional Systems, Warren, NJ) (Figure 31.1, right). The first coronary Palmaz-Schatz stent was placed in a patient by Eduardo Sousa in São Paulo, Brazil in 1987 with a US pilot study started in 1988.
 Figure 31.1 Left. The Gianturco-Roubin Stent. Stainless steel sutures were wound around a cylindrical rod using pegs to shape the wire, resulting in a clamshell design. Right. The Palmaz-Schatz Stent. Note the articulation between the two slotted tubes. |
of the Palmaz-Schatz stent by the FDA in 1994. Long-term follow-up up to 15 years has subsequently demonstrated few late clinical or angiographic recurrences from years 1 to 5 after coronary stent implantation,8,9 with slight and progressive decrements in luminal size thereafter extending beyond 10 years.10 The mechanisms of this late progression of disease are not entirely known, but have been hypothesized to be related to the development of new atherosclerosis within the originally stented segment rather than clot formation, as overall stent thrombosis rates have remained low (1.5% at 15 years).10
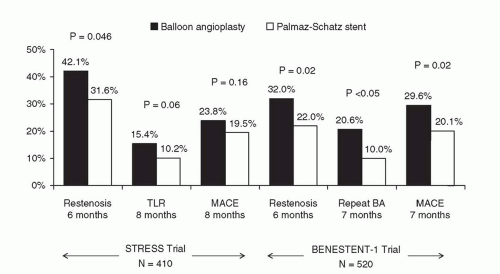 Figure 31.2 Results of STRESS and BENESTENT-1 landmark trials of the Palmaz-Schatz stent, which provided the evidence base for FDA approval of the Palmaz-Schatz stent for the prevention of restenosis in de novo lesions. BA, balloon angioplasty; TLR, target lesion revascularization; MACE, major adverse cardiac events. |
details. In recent years, the importance of the stent delivery system to device profile, flexibility, and trackability around tortuous and calcific coronary vessels has received increasing appreciation. For balloon-expandable stents, the stent must be tightly crimped to the delivery balloon to avoid dislodgment, and the overhang of the balloon beyond the ends of the stent should be minimized (<1 mm) to avoid vessel trauma outside the stent margins. Stent delivery balloons must be able to withstand high pressures (>18 atm) without rupture, and should take into account a balance between deliverability versus a desire for low compliance to facilitate predictable sizing and avoid excessive growth outside the stent edges.
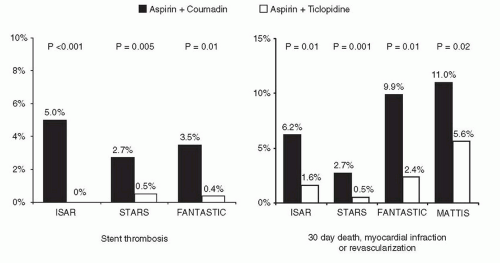 Figure 31.3 Benefits of dual antiplatelet therapy in reducing clinical events post stenting. Shown are the results from four landmark trials demonstrating the efficacy of antiplatelet (over antithrombotic) therapy. |
struts may be associated with reduced neointimal hyperplasia and lower rates of restenosis,20 in addition to inherently less thrombogenicity.21
Table 31.1 Stent Coatings Designed to Reduce Thrombogenicity | ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||
regarding the long-term safety of DES and the requirement for extended duration dual antiplatelet therapy have led to a renewed interest in biocompatible stent coatings. A number of novel stent coatings are currently under investigation. Additionally, covered stents (metallic stents covered by a distensible microporous PTFE membrane) are of unquestioned clinical utility in treating life-threatening perforations (see Chapter 4). They are also used for excluding giant aneurysms, pseudoaneurysms, or clinically significant fistulae.
performed today, and balloon angioplasty alone is reserved for cases where stents cannot be delivered, where stents are too big for the target lesion, or for rare niche indications (e.g. ostial side branch disease at a bifurcation, some cases of instent restenosis, or cases where patients cannot tolerate the antiplatelet regimens required after stent implantation).
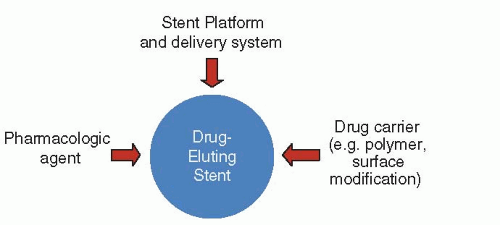 Figure 31.4 Components of drug-eluting stents. |
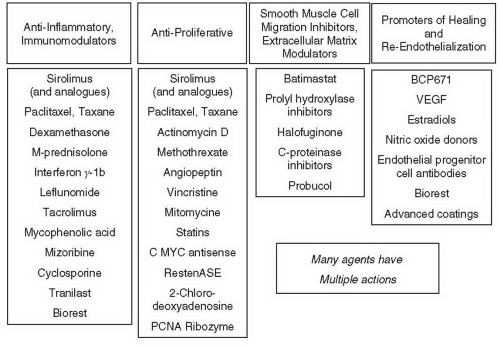 Figure 31.5 Potential antirestenotic agents for use with drug-eluting stents. |
itself is not well known, but in animal models these effects can be attenuated by modification of the polymer vehicle.53 It is believed that inflammation and delayed endothelialization play a role in the development of late stent malapposition, aneurysm formation, stent thrombosis and restenosis.50,54,55 For these reasons, there has been great interest in developing inert and biocompatible polymers, bioabsorbable/biodegradable polymers, and even polymer-free DES.
Table 31.2 Generational Classification of Drug-Eluting Stents | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
leading to its approval in Europe in 2002 and in the United States in 2003. Production of this stent was recently halted, but some description of the technology and initial studies that led to device approval is of historical interest, as the introduction of this stent ushered in the DES era of interventional cardiology. Sirolimus (rapamycin) is a highly lipophilic, naturally occurring macrocyclic lactone, which was first isolated from Streptomyces hygroscopicus found in a soil sample from Easter Island (also known as Rapa Nui) and was initially developed as an antifungal agent. Shortly thereafter, it became apparent that this agent also was a potent immunosuppressive, and was initially approved by the FDA (as Rapamune) for prevention of renal transplant rejection in 1999. The primary mechanism of action of inhibition of neointimal hyperplasia in sirolimus is thought to be related to its ability to bind to FKBP-12 in cells; the sirolimus-FKBP-12 complex then binds to and inhibits activation of mTOR, preventing progression in the cell cycle from the late G1 to S phase.43 Sirolimus has been demonstrated to have a marked effect on suppression of neointimal hyperplasia with low toxicity following sirolimus-eluting stent (SES) implantation in initial small and large animal studies.56,57
of 2.5 to 3.5 mm and lesion lengths of 15 to 30 mm. The primary endpoint, the rate of target vessel failure (TVF, a composite of cardiac death, myocardial infarction [MI], or target vessel revascularization [TVR]) at 9 months, was markedly lower among SES-treated patients (8.6% versus 21.0%, P < 0.001) (Figure 31.7). Additionally, SES resulted in a 60% to 80% relative reduction in composite adverse events in all examined subgroups in the trial. Among the 703 patients in whom 8-month routine angiographic follow-up was performed, mean in-stent late loss was markedly lower with SES (0.17 mm versus 1.00 mm, P < 0.001). By IVUS, the in-stent percent volumetric obstruction at 8 months was reduced from 33.4% with the Bx Velocity to 3.1% with the SES (P < 0.001), although late stent malapposition was present in 9.7% of Cypher™ SES patients versus 0% of Bx Velocity patients (P = 0.02).
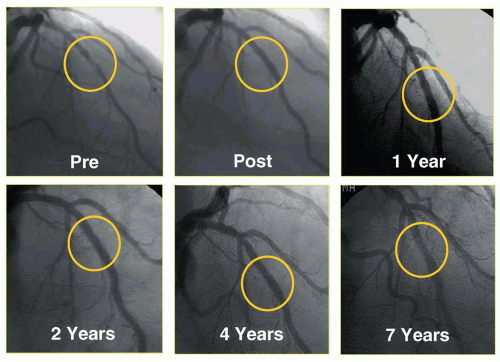 Figure 31.6 Seven-year follow-up of one of the initial sirolimus-eluting stent implantations from Institute Dante Pazzanese of Cardiology in São Paulo, Brazil, demonstrating sustained patency of the initially stented segment. |
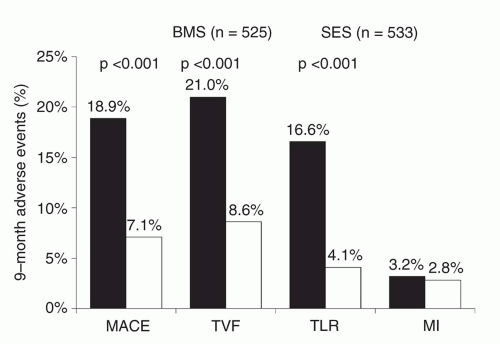 Figure 31.7 Primary results of the SIRIUS trial, the pivotal approval trial of the sirolimuseluting stent, demonstrating superiority of the sirolimus-eluting stent in reducing restenosis-related endpoints. SES, sirolimus-eluting stent; BMS, bare-metal stent; MACE, major adverse cardiac events; TVF, target vessel failure; TLR, target lesion revascularization; MI, myocardial infarction. |
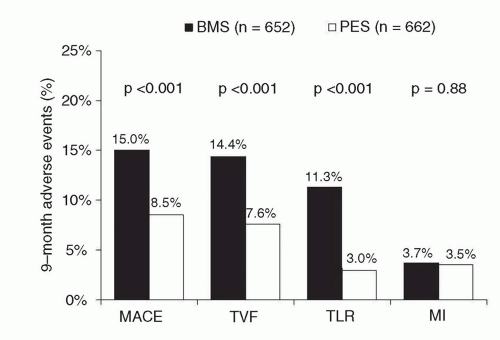 Figure 31.8 Primary results of the TAXUS-IV trial, the pivotal approval trial of the paclitaxel-eluting stent, demonstrating superiority of the paclitaxel-eluting stent in reducing restenosis-related endpoints. PES, paclitaxel-eluting stent; BMS, bare-metal stent; MACE, major adverse cardiac events; TVF, target vessel failure; TLR, target lesion revascularization; MI, myocardial infarction. |
Table 31.3 Randomized Controlled Trials of Everolimus-Eluting Stents | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(nearly 50% of whom presented with acute coronary syndromes) were randomized to ZES(E) versus SES. In this trial, treatment with ZES(E) was associated with higher rates of 9-month major adverse cardiac events (MACE: cardiac death, MI, or TVR: 6% versus 3%, P = 0.0002), as well as endpoints of MI, stent thrombosis, and TLR, differences which persisted at 18 months (with the exception of stent thrombosis). The ISAR-TEST-2 trial was a three-way 1:1:1 randomized trial in 1,007 patients of an investigational combination sirolimus/probucol-eluting stent versus ZES(E) versus SES.111,112 Compared to SES, ZES(E) resulted in higher rates of late loss, angiographic restenosis (the primary endpoint), and TLR at 6 to 8 months, with similar rates of death, MI, and stent thrombosis. A larger study, the ZEST trial, randomized 2,645 patients with simple and complex coronary artery disease to ZES(E), SES, or PES.113,114 In this trial, while SES demonstrated the lowest degree of late loss and binary restenosis, ZES(E) was intermediate between SES and PES with respect to rates of MACE, TVR, and TLR. There were no significant differences in the 2-year rates of death, MI, or stent thrombosis between the two stents.
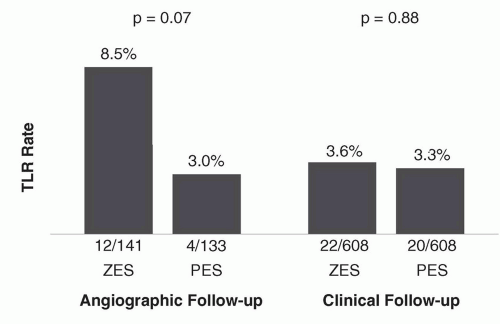 Figure 31.9 Rates of target lesion revascularization in the ENDEAVOR IV trial according to the performance of angiographic follow-up. The differences between stents are minimized among the majority of patients undergoing clinical follow-up alone. TLR, target lesion revascularization; ZES, zotarolimus-eluting stent; BMS, bare-metal stent. |
stents (8.1% versus 8.2%, P = 0.94), with no observed differences in other clinical endpoints, including stent thrombosis (definite/probable: 0.9% for ZES(R) versus 1.2% for EES).
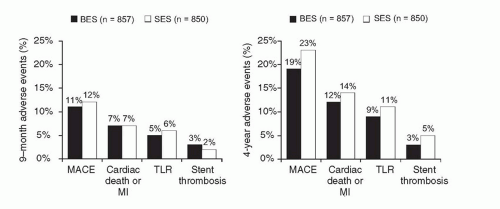 Figure 31.10 Principal clinical endpoints at 1 year (left) and 4 years (right) from the randomized all-comers LEADERS trial of a biolimus A9-eluting stent compared to a sirolimus-eluting stent. BES, biolimus A9-eluting stent; SES, sirolimus-eluting stent; MACE (major adverse cardiac events) denotes cardiac death, myocardial infarction (MI), or clinically indicated target vessel revascularization; stent thrombosis refers to Academic Research Consortium (ARC) definite or probable events. |
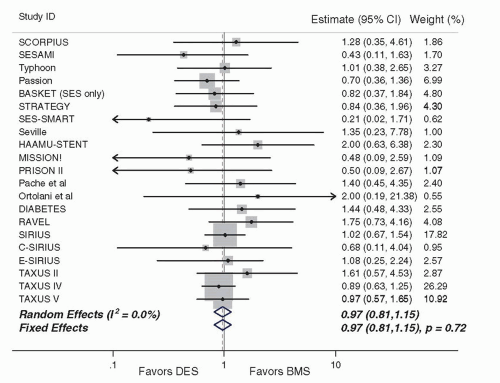 Figure 31.11 Mortality in randomized trials comparing drug-eluting stents to bare-metal stents (from Kirtane et al., Circulation 2009), demonstrating similar overall mortality of both stent types. DES, drug-eluting stent; BMS, bare-metal stent. |
included trials.127 In this analysis of 38 trials including data from 18,023 patients, TLR was lower with SES and PES compared to BMS, with similar mortality among patients treated with SES, PES, and BMS. In this analysis, a reduction in the hazard of MI was observed with SES compared to both BMS (hazard ratio [HR] 0.81, 95% credibility interval 0.66 to 0.97, P = 0.030) and PES (HR 0.83, 0.71 to 1.00, P = 0.045).
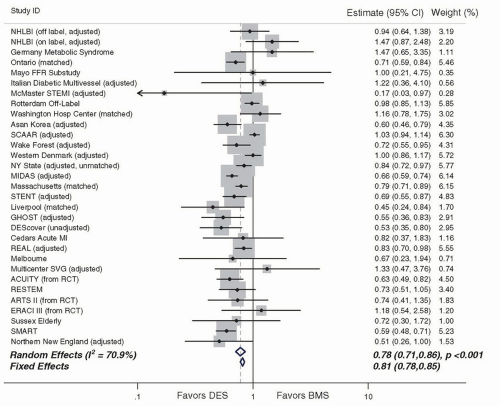 Figure 31.12 Mortality in observational studies comparing drug-eluting stents to bare-metal stents (from Kirtane et al., Circulation 2009), demonstrating a reduction in mortality with drug-eluting stents. DES, drugeluting stent; BMS, bare-metal stent. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


