Chapter 26 Electrophysiological Evaluation of Recurrent Ventricular Tachycardia
Introduction
Ventricular tachycardia (VT) is typically an ominous sign in the setting of structural heart disease. Its evolution to ventricular fibrillation (VF) and sudden cardiac death (SCD) has become increasingly recognized. Patients at particularly high risk are those with chronic coronary artery disease (CAD) with prior myocardial infarction (MI) and marked left ventricular (LV) systolic dysfunction.1 In the post-thrombolytic era, the incidence of sustained VT after MI is still approximately 2% to 4% in the first 48 hours and then stabilizes to about 0.5% per year thereafter. As many as 5% to 10% of the patients will experience VF or SCD that is usually preceded by sustained VT.2,3 An estimated 40% of individuals who have an implantable cardioverter defibrillator (ICD) placed for an index event of sustained VT will have recurrent VT.4,5 However, all VTs are not equal by nature; some can occur in the absence of structural heart disease and have a very benign prognosis. The disease states associated with VT and the management of VT will be addressed in greater length in other chapters of this text. This chapter will delineate the approach to the electrophysiological evaluation of recurrent VT.
Mechanisms
Abnormal Automaticity
Normal automaticity refers to reliable depolarization and impulse formation of pacemaker cells. Such cells in the ventricle normally provide escape rhythms of less than 50 beats/min. VT caused by abnormal automaticity may occur when ventricular myocytes generate impulse formation at an accelerated rate compared with the normal rate because of an altered threshold for sodium (Na+) influx into the cells. This mechanism is thought to underlie accelerated idioventricular rhythms in the setting of acute hypokalemia, hypomagnesemia, cocaine intoxication, focal inflammation, acute myocarditis, or ischemia.5 In the absence of such derangements, VTs of this type typically are seen in children or young adults.
Triggered Activity
Action potentials may occasionally propagate afterdepolarizations caused by oscillations in the membrane potential. Such triggered activity may either occur as early afterdepolarizations (EADs) or delayed afterdepolarizations (DADs). EADs occur during phase II or III of the action potential, are more likely to occur during bradycardia, and are implicated in torsades de pointes induced by drugs or electrolyte abnormalities and in some variants of congenital long QT syndrome (LQTS). DADs occur during phase IV of the action potential, are more tachycardia dependent, and are believed to be responsible for ventricular arrhythmias occurring in digitalis intoxication and outflow tract VTs.5 The mechanism may also play a role in VT in the setting of acute MI.5
Re-entry
Re-entry is, by far, the most common mechanism of VT associated with structural heart disease. Clinical characteristics include spontaneous induction by premature ventricular complexes with usually abrupt initiation and termination. It requires tissue that can conduct unidirectionally with either fixed or functional block as well as a region of relatively slow conduction that permits recovery of previously depolarized tissue and a circus rhythm to propagate. The mechanism underlying most VTs in the setting of structural heart disease is re-entry caused by a scar. Although a myocardial scar or functional block caused by a nonischemic insult can serve as the pathophysiological substrate, chronic CAD with prior infarct, particularly in the setting of compromised LV function, is the most common underlying pathology.1
Noninvasive Assessment
Surface Electrocardiogram
As in the face of any wide-complex tachycardia, a supraventricular etiology, with intrinsic bundle branch block, aberrancy, or antegrade accessory pathway conduction, should first be ruled out. The baseline electrocardiogram (ECG) can be very helpful in the initial assessment of presumed VT.5 The presence of a bundle branch block or manifest pre-excitation at baseline that is identical to that seen during clinical tachycardia speaks against VT as a diagnosis. Importantly, even minor differences in the wide QRS complex tachycardia compared with the sinus rhythm recording suggest a ventricular origin. The presence of Q waves in contiguous leads or ST elevations in the absence of acute MI suggests prior MI and aneurysm with a scar, respectively, and strongly suggests VT, especially if the infarct site is consistent with the presumed VT exit (see below). The presence of a widened QRS at baseline, which is a marker of His-Purkinje disease, suggests a predisposition for bundle branch re-entrant or fascicular VT. If a 12-lead ECG of the clinical arrhythmia is available, the presence of ventriculoatrial dissociation or capture or fusion beats is diagnostic for VT. Additional axis and morphology characteristics may also be present that may point toward VT as the diagnosis.3,6
If a 12-lead ECG has been obtained during VT, much additional information may be gleaned from it. First, the “clinical” VT will be apparent, which will be of great assistance during electrophysiological study prior to ablation. Pacemapping strategies will be greatly assisted by this in the setting of poorly hemodynamically tolerated or noninducible VTs and substrate-based ablation (discussed in later chapters). Knowing which of many potentially inducible VTs during electrophysiological study are pertinent also will ensure that those VTs are targeted during the ablation procedure. Finally, a likely “site of origin,” or at least close approximation of the VT exit from a larger macro–re-entrant circuit, can often be determined.7
A general strategy should begin with determining the bundle branch morphology. A left bundle branch block (LBBB) pattern, defined by presence of a terminal S wave in the QRS complex in lead V1, indicates either a right ventricular (RV) origin or a septal LV origin. A right bundle branch block (RBBB) pattern, defined by the presence of a terminal R in V1 almost uniformly indicates an LV origin. The frontal plane axis should then be examined. In the inferior leads, a positive QRS axis will indicate a superior (thus inferiorly directed) VT exit, and a negative axis will indicate an inferior one. Transition in the precordial leads, defined by the first precordial lead where the R is greater than S, indicates how basal (transition ≤ lead V2) or apical (negative throughout precordium) is the VT circuit exit or focus.7 This strategy, coupled with intracardiac mapping (described below) works well in almost all cases except for VT occurring in the presence of a large apical infarction, in which septal versus lateral exits are difficult to distinguish.8 Assessing the timing of the surface QRS onset to the RV apical intracardiac recording during VT suggests a lateral origin if the time exceeds 100 ms.8
The above localization strategy was developed in patients with re-entrant VT caused by chronic CAD and prior infarction but can be applied to other forms of VT as well. Here, clinical history and pattern recognition can be additionally helpful. In a young patient with no known heart disease or evidence for CAD, VT with LBBB and tall, monomorphic R waves in leads II, II, and aVF strongly suggest an outflow tract origin and idiopathic VT. A young, athletic person with several premature ventricular contractions (PVCs) or VT morphologies localized to the RV should raise suspicion of arrythmogenic right ventricular cardiomyopathy (ARVC), particularly in the presence of ε-waves or T-wave inversions in the early precordial leads, V1 to V3, or other surface ECG clues during sinus rhythm. It is important to note, however, that re-entrant VT associated with nonischemic cardiomyopathy typically originates near the peripulmonic, aortic, superior or all of these mitral valves and can mimic right ventricular outflow tract/left ventricular outflow tract (RVOT/LVOT) morphology.5
Imaging
Noninvasive imaging is often much more sensitive than the surface ECG in assessing for presence of structural heart disease and, in particular, for a scar or ischemia. This can usually be easily accomplished by traditional methods of echocardiography or nuclear scintigraphy. Magnetic resonance imaging (MRI) is a generally less accessible but often more sensitive tool for detecting both LV and RV abnormalities.9 The presence of an ICD or pacemaker has long been viewed as a contraindication for performing an MRI, but experiences at several centers, including those of the authors of this chapter, have shown that it can be done safely.10 The presence of a sessile intracardiac thrombus should be ruled out before any invasive evaluation or intervention is attempted in the presence of significant structural heart disease, given the high risk for thromboembolism and stroke.
Electrogram Information from the Implanted Cardioverter Defibrillator
For patients with an ICD, the VT information stored on the device should be carefully reviewed. The number of events, both nonsustained ones and those resulting in therapy, reflects the arrhythmia burden and should determine the urgency with which subsequent medical care is executed. This includes initiation of antiarrhythmic therapy and catheter ablation.1 Device-recorded intracardiac electrograms (EGMs) may be especially helpful in determining if catheter ablative therapy may be required; if it is, EGMs will help target the ablation site if a surface 12-lead ECG of the clinical VT is not available. Events predominated by polymorphic VT without clear monomorphic PVC trigger, for instance, may be less amenable to ablation unless a reproducible trigger is evident. Depending on the stability of the patient’s condition, noninvasive programmed stimulation (NIPS) testing through the device could be considered to find out if the intracardiac EGM of any induced VT resembles those stored from spontaneous clinical events so that the occurrence of clinical VT may be identified on 12-lead ECG.
Invasive Assessment of Ventricular Tachycardia
Initiating Ventricular Tachycardia
A hallmark of re-entrant VT is the ability to induce it and terminate it by ventricular programmed electric stimulation (PES) in the electrophysiological laboratory. Because of changes in refractory periods and tissue conduction velocities, along with variations in heart rates, re-entry is favored by fast heart rates and sudden changes in rate. A critical basic pacing rate in PES, therefore, is often needed to initiate a re-entrant tachycardia in a given patient, and several different basic pacing drive cycle lengths with extrastimuli should be used to try to induce it.7 Notably, triggered VTs also can be occasionally initiated with ventricular PES; thus, induction with PES does not exclude the latter as a mechanism for VT.7 In inducing VT with PES, it is important to attempt stimulation from additional ventricular sites other than the RV apex. Additional stimulation from sites such as the RVOT or the LV may be required to initiate a monomorphic, sustained VT in 10% to 20% of patients in whom stimulation from the RV apex is ineffective.7 Frequently, VT with a RBBB pattern will best be initiated by stimulating from the lateral LV.
Methods for PES vary significantly. Pacing for eight beats with basic drive cycle lengths of 600 and 400 ms followed by the introduction of one to four extrastimuli is generally the most accepted protocol. The ability to induce sustained monomorphic VT (MMVT) increases successively with the number of extrastimuli, up to three. MMVT can be induced in 96% of those with sustained clinical VT and in 75% of those who have survived sudden arrhythmic cardiac death.7 Beyond three extrastimuli, additional yield is minimal; sensitivity and inducibility increase marginally, but with a significant decrease in specificity as less stable and more nonclinical VTs, polymorphic VTs (PMVTs), or both are induced.7 In the majority (80% to 85%) of patients, clinical VT can be induced in this fashion; however, occasionally, induction is more effective when the basic “drive” is provided by sinus rhythm, with extrastimuli introduced via pacing. If a specific VT is identified, the introduction of additional extrastimuli may be used for induction, recognizing the potential for the induction of less clinically relevant arrhythmias. On occasion a short-long-short stimulation sequence may be advantageous. This stimulation has been described for the induction of bundle branch re-entry but can be used for the induction of other VTs associated with structural heart disease that are not induced with a more standard stimulation protocol.7
Induction of VT with burst pacing or sympathetic stimulation, for instance, with isoproterenol infusion or stress testing, is much more likely to occur with triggered arrhythmias. Interestingly, atrial burst pacing is somewhat more likely to induce outflow tract tachycardias compared with ventricular burst pacing.3
VT caused by abnormal automaticity is not affected by PES, either in terms of initiation or termination; the inability to induce clinical VT with PES, as described above, suggests automaticity as a mechanism. These VTs are affected by basic heart rate and are typically elicited during bradycardia.7 Some are enhanced by the infusion of isoproterenol.
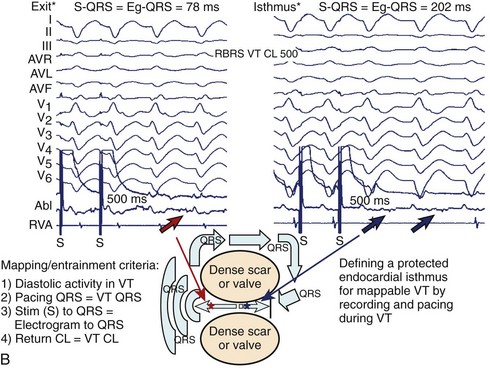
Certainly, the goal of these procedures is to reproducibly initiate VT and to perform maneuvers that lend support to its underlying mechanism. If the diagnosis of VT is not certain, confirming it in the electrophysiological laboratory is fairly straightforward. The diagnosis of VT can be established with the mode of arrhythmia induction and the presence of atrial and ventricular dissociation at the onset of VT elicited with pacing maneuvers. Atrial stimulation can be used during a wide complex tachycardia that is suspected of being antidromic SVT over a bypass tract when the His atrial activity is refractory to confirm atrial participation in the circuit. As alluded to earlier, re-entrant VT can be initiated and terminated by ventricular PES. Its other hallmarks are (1) an inverse relationship between the coupling interval of the ventricular extrastimulus and the return cycle length of the tachycardia; (2) the ability to entrain the VT with progressive fusion with shorter pacing cycle lengths; and (3) the ability to demonstrate entrainment with concealed fusion at isthmus sites.3
Mapping Strategies: Hemodynamically Stable, Re-entrant Ventricular Tachycardia
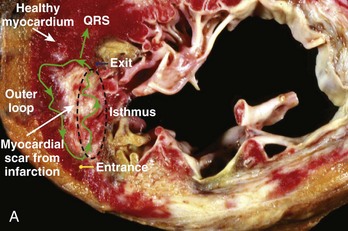
Hemodynamically stable, relatively slow VT that is easily inducible and stable in response to pacing comprises a small minority of re-entrant VTs, especially in the setting of ischemic heart disease. However, when re-entrant VT is present, detailed activation and entrainment mapping can be performed. The goal of both is to identify the critical elements of the re-entrant circuit that are protected by anatomic or functional boundaries (a dense scar or valvular structures) and which would therefore be easiest to successfully target with ablation. The surface ECG recording of VT typically represents the wavefront as it exits the scarred area and begins to depolarize the healthy myocardium. The “exit” is frequently at the end of the most critical portion (isthmus) of the circuit and is located at the border of a scar. However, it is possible for the isthmus to be quite far from the edge of the scar and the wavefront to activate the diseased myocardium for several centimeters before engaging the normal myocardium. Once the wavefront encounters the normal myocardium, it then propagates away from this site to depolarize the rest of the ventricles as well as to complete the depolarization of other portions of the circuit, including the outer scar border (outer loop) and the “entrance” to the isthmus region (Figure 26-1, A). Occasionally, the wavefront may propagate through a path within the scar or area of functional block (inner loop) that is not critical to the maintenance of the circuit.11
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


