Depression and Coronary Heart Disease
During the past 2 decades, a rapidly accumulating body of neurobiologic knowledge has catalyzed fundamental changes in how we conceptualize depressive symptoms in patients with atherosclerotic vascular disease. Buttressing older investigations of type A personality, hostility, and styles of psychological coping are vital, ongoing scientific developments that flow from an increased understanding of the interplay among the immune system, vasculature, and brain. Indeed, the evolving understanding of the interplay between the neurobiology of the stress response, neuroendocrine function, and immunoinflammatory vascular pathways and resulting neurobehavioral symptoms of the heart patient represent a quintessential mind-body interface.
Epidemiology
Depressive syndromes and major depression are exceedingly common. The most recent comprehensive studies conducted in the United States, the National Comorbidity Study Replication (NCS-R) and its predecessor, the National Comorbidity Study, reported lifetime and 12-month prevalence rates of major depression1 of 16% and approximately 7%, respectively. Minor depressive disorder (depressive symptoms subthreshold in severity compared with major depression and dysthymia) is also common in the community2 and in primary care clinics.3 The Epidemiologic Catchment Area Study of more than 18,500 individuals reported the lifetime prevalence rate of subthreshold depressive symptoms to be 23% in comparison with 6%, the sum of the prevalence rates of major depression and dysthymia.2 Not only do depressed patients experience greater difficulties in problem solving and coping with challenges, but depression adversely affects adherence to medical therapy4 and rehabilitation,5 as well as the quality of medical care received.6 Minor depressive disorder is also associated with significant functional impairment and substantial increases in healthcare utilization.2 Of note is that point prevalence rates in primary care outpatients range from 2% to 16% for major depression and 9% to 20% for all depressive disorders.7 The rates are even higher among medical inpatients: 8% for major depression and 15% to 36% for all depressive disorders8 (Table 96–1).
| Major depressive disorder |
| • Five (or more) of the following symptoms have been present during the same 2-wk period and represent a change from previous functioning; at least one of the symptoms is either (1) depressed mood or (2) loss of interest or pleasure. |
| (1) Depressed mood |
| (2) Markedly diminished interest or pleasure |
| (3) Significant weight loss or weight gain, or decrease or increase in appetite |
| (4) Insomnia or hypersomnia |
| (5) Psychomotor agitation or retardation (observable by others) |
| (6) Fatigue or loss of energy nearly every day |
| (7) Feelings of worthlessness or excessive or inappropriate guilt |
| (8) Diminished concentration or indecisiveness |
| (9) Recurrent thoughts of death (not just fear of dying) or suicide |
| • The symptoms cause clinically significant distress or impairment in social, occupational, or other important areas of functioning. |
| • The symptoms are not because of the direct physiologic effects of a substance or a general medical condition. |
| • The symptoms are not better accounted for by bereavement. |
| Dysthymic disorder |
| A. Depressed mood for most of the day, for more days than not, for at least 2 years |
| B. Presence, while depressed, of 2 (or more) of the following: |
| (1) Poor appetite or overeating |
| (2) Insomnia or hypersomnia |
| (3) Low energy or fatigue |
| (4) Low self-esteem |
| (5) Poor concentration or difficulty making decisions |
| (6) Feelings of hopelessness |
| C. The disturbance is not better accounted for by chronic major depressive disorder. |
A number of longitudinal, community-based epidemiologic studies have assessed depressive symptoms in predicting coronary heart disease (CHD). Assessments of this type typically are added to large, multiple risk factor studies in which population-based samples are prospectively followed.9 The advantage of using dimensional measures of depression (rather than a categorical diagnosis of major depression) lies in the increased statistical power that allows these studies to detect smaller effects. A meta-analysis by Wulsin and Singal10 documented that, even after controlling for traditional risk factors for CHD (eg, physical inactivity, nicotine use, hypertension), the presence of depressive symptoms is a significant independent risk (relative risk [RR] 1.64; 95% confidence interval: 1.41-1.90) for the onset of CHD. Moreover, studies revealing increasing graded relative risks for heart disease in persons with increasing severity of depression symptoms11,12 provide further evidence that depression plays a causal role in CHD.
Since the 1960s, multiple studies, both cross-sectional and longitudinal in design, have examined the association of cardiovascular disease (CVD), especially CHD and congestive heart failure (CHF), with depressive symptoms and major depression. Despite the potential methodologic weaknesses of some of the studies, point prevalence rates of depression are relatively consistent, ranging from 14% to 23%.13 Furthermore, evidence suggests that major depression, aside from acting as an independent risk factor for the development of CHD, is associated with drastically elevated morbidity and mortality for patients across the spectrum of CHD, including patients with stable CHD,14 after an index myocardial infarction (MI), and in patients with atrial fibrillation or heart failure.15 Both major depressive disorder (MDD) and elevated depressive symptoms are associated with a poorer prognosis in patients with CHD.16-20 Furthermore, there are earlier and more severe cardiac events with increasing severity of comorbid depressive symptoms.16,18
The negative impact of depression in patients with CVD was initially highlighted by the seminal studies of Frasure-Smith and colleagues in the 1990s. These researchers at the Montreal Heart Institute discovered that patients with major depression after an index MI suffered three- to five-fold increased odds of dying within 6 and 18 months post-MI,21 even after controlling for prognostic factors such as left ventricular dysfunction and previous MI. More recent studies are concordant with these results.22,23 Indeed a meta-analysis of 22 studies revealed that post-MI depression is associated with a 2- to 2.5-fold increased risk of a poor outcome, including mortality and future cardiovascular events.24 In fact, the severity of depressive symptoms has as great a detrimental impact on survival as does left ventricular dysfunction or diabetes.13
Depression also exerts a negative impact on patients with CHF25-27 or recent coronary artery bypass grafting (CABG) surgery.28,29 In a large prospective study of 817 patients undergoing CABG with a 12-year follow-up period, Blumenthal and colleagues29 showed that patients with moderate to severe depressive symptoms before surgery suffered a greater than two-fold risk of death post-CABG when compared with nondepressed patients, even after controlling for ethnicity, diabetes, obesity, left ventricular ejection fraction, history of cigarette smoking, number of grafts, or history of MI.
Taken together, the concatenation of data strongly support the notion that depression is an independent risk factor in the pathophysiologic development and progression of CVD, rather than merely a secondary emotional response to cardiovascular illness. The significance of these findings are such that the American Heart Association issued recommendations for screening, referral, and treatment of depression in patients with CHD.30 Recognition and treatment of major depression is crucial for all patients, but especially for patients after an MI, who are 3 times more likely to develop depression than the general population. Furthermore, depression remains an independent risk factor for death 5 years after an acute MI.20
The Heart and Soul Study was specifically designed to determine why depression is associated with increased cardiovascular events in patients with stable CHD and found that depressive symptoms are associated with patient-reported health status, quality of life, and overall health, but not with the traditional measures of cardiac health—ejection fraction and ischemia.31 A follow-up to the Heart and Soul Study further suggests that the association between depressive symptoms and adverse cardiovascular events is largely explained by physical inactivity, in addition to other behavioral factors.32 Though the directionality of this relationship has not yet been established (likely bidirectional), it is potentially modifiable with interventions targeted toward increased activity in this patient population.
Pathophysiology
Advances in biologic psychiatry have included the discovery of numerous neurochemical, neuroendocrine, and neuroanatomic alterations in unipolar depression, some of which likely contribute to the increased vulnerability of depressed patients to CVD. These biologic changes include immune-system activation with increased secretion of proinflammatory cytokines, endothelial dysfunction, hypothalamic-pituitary-adrenocortical (HPA) system and sympathoadrenal hyperactivity, diminished heart rate variability, alterations in platelet receptors and/or reactivity, and ventricular instability and myocardial ischemia in reaction to mental stress (Fig. 96–1). Evidence further suggests that the relationship between inflammation and autonomic dysfunction is more robust among depressed patients than nondepressed counterparts.33 And most recently, a candidate gene study suggested that genetic variation related to endothelial dysfunction is predictive of depressive symptoms in patients with cardiovascular disease.34 Taken together, these findings support the premise that genetic, biologic, and behavioral factors interact in a complex and not as yet fully defined, multidirectional fashion that ultimately links depression and cardiovascular disease.
Figure 96–1.
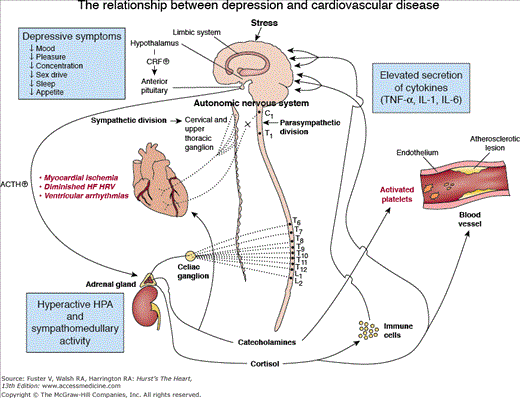
Hypothetical schema of pathophysiologic findings associated with depression that probably contributes to increased susceptibility to cardiovascular disease. Autonomic nervous system innervation of the heart via the parasympathetic vagus (X) nerve and sympathetic (postganglionic efferents from the cervical and upper thoracic paravertebral ganglia) nerves is shown. ACTH, corticotropin; CRF, corticotropin-releasing factor; HRV, heart rate variability; HPA, hypothalamic-pituitary-adrenocortical axis; IL-1, interleukin-1; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α. Reproduced with permission from Arch Gen Psychiatry. 1998;55:583. Copyright © 1998, American Medical Association.
Activation of inflammatory pathways has been implicated in the pathogenesis of both cardiovascular illness and mood disorders (Table 96–2). It has been clearly established that atherosclerosis is primarily an inflammatory disease35,36 (see Chaps. 52 and 53). Similarly, there is now unequivocal evidence that depression is associated with an activated innate immune response, although it is unclear whether this activation is the etiology of depression or a consequence of the mood disorder.37
| Depression | Cardiovascular Disease | Role in Inflammation |
|---|---|---|
| CRP27-29 | CRP30-32 | Amplifies inflammatory and procoagulant responses33 |
| Induces expression of cellular adhesion molecules, which mediates adhesion of leukocytes to the vascular endothelium33 | ||
| Induces monocyte expression of tissue factor33 | ||
| IL-1β34,35 | IL-1β36 | Induces expression of cellular adhesion molecules, which mediates adhesion of leukocytes to the vascular endothelium33 |
| Augments MMP expression, which is involved in the degradation of the subendothelial basement membrane37 | ||
| IL-628,29 | IL-636,38,39 | Principal procoagulant cytokine33 |
| Stimulates hepatic production of CRP and fibrinogen40 | ||
| IL-741 | Regulates T-cell homeostasis42 | |
| Stimulates monocyte production of chemokines41 | ||
| IL-834 | IL-836 | |
| Interferon-γ43 | Interferon-γ30 | Augments synthesis of TNF-α and IL-144 |
| TNF-α28,34 | TNF-α36 | Induces expression of cellular adhesion molecules, which mediates adhesion of leukocytes to the vascular endothelium33 |
| Augments MMP expression, which is involved in the degradation of the subendothelial basement membrane37 | ||
| Nuclear factor-κB45 | Induces transcription of the VCAM-1 gene45 | |
| Fibrinogen46 | Fibrinogen40 | Key component of the coagulation cascade |
| Soluble CD-40 ligand | Augments MMP expression, which is involved in the degradation of the subendothelial basement membrane37 | |
| VCAM-147 | Mediates early adhesion of mononuclear leukocytes to arterial endothelium37 |
A key component of the inflammatory response involves the secretion of proinflammatory cytokines by activated cells such as endothelial cells, fibroblasts, macrophages, and monocytes. Cytokines coordinate immune responses and provide communication between multiple sites, including sites of infection, atherosclerotic plaques, adipose tissue, the liver, and the CNS. In addition to their biologic activity, acute-phase proteins also induce neurocognitive and behavioral changes known as sickness behavior. Sickness behavior, a constellation of nonspecific signs and symptoms that accompanies the physiologic response to infection and inflammation, includes fatigue, anorexia, anhedonia, decreased psychomotor activity, and disappearance of body-care activities,37,38 all of which overlap with symptoms of major depression.
Depression, in otherwise medically healthy individuals, is associated with increased production of proinflammatory cytokines, other cytokines, and acute-phase reactants, including interleukin (IL)-1β, IL-2, IL-6, IL-8, interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), monocyte chemotactic protein-1 (MCP-1), fibrinogen, and C-reactive protein (CRP).37,39-42 There is also evidence that plasma concentrations of IL-1β and IL-6 correlate strongly with depression severity.37,43 In the multiple studies examining the relationship between CRP and depression, the evidence supports a definitive association. In an analysis of 4218 individuals older than 65 years and free of cardiovascular disease, investigators evaluated the presence of exhaustion and depressive symptoms with respect to levels of CRP, albumin, white blood cells (WBCs), various coagulation factors, and fibrinogen. Depressive symptomatology was associated with elevated markers of low-grade inflammation, including CRP and fibrinogen.42 This relationship held true after adjusting for some control variables (age, sex, race, height, weight, diabetes mellitus, smoking, and systolic blood pressure), but not others (grip strength, 15-ft walk time, and activity level). Similarly, analysis of data from the ATTICA study,44 a health and nutrition survey conducted in Greece with a total of 4056 individuals available for potential inclusion, including 453 men (18-89 years old) and 400 women (18-84 years old), showed that scores from the Zung depression scale correlated in a dose-response relationship, with elevated levels of CRP, WBC count, and fibrinogen in both sexes.45 Again, subjects in this study were all cardiovascular disease-free. Douglas and colleagues46 also found that depression scores correlated positively with serum concentrations of CRP in active duty US Army personnel undergoing routine physicals, but not if adjusted for body mass index (BMI). Other investigators have obtained similar results linking BMI with depression and levels of CRP.
There are sex differences with respect to depression and elevated cytokine levels. Ford and Erlinger39 studied 6914 young adults between the ages of 18 and 39 years using data from the Third National Health and Nutrition Examination Survey. After adjustment for numerous potential confounders, there was a significant relationship between a lifetime history of major depression and elevated CRP levels in men, but not in women. This sex difference with respect to elevated CRP levels and depression is particularly interesting given that both men and women have a similar RR of CHD related to depression.16 An earlier study by Danner and colleagues47 evaluated the same patient population as the Ford group with similar results, suggesting that a sustained inflammatory response can result after a depressive episode.
Further supporting this notion of a depression-induced sustained inflammatory response is a study of 119 older adults, immediately before and 2 weeks after, an annual influenza vaccination.48 Among all participants, those with more depressive symptoms at baseline had higher levels of IL-6 before vaccination and experienced a subsequent increase in IL-6 after vaccination. Those patients with fewer depressive symptoms had lower baseline levels of IL-6 and exhibited little change in IL-6 levels after vaccination.
Exogenous cytokine therapy has also been shown to be associated with the onset of behavioral changes, including major depressive episodes. IL-2 and IFN-α are both used in the treatment of melanoma and hepatitis C. In individuals free of depression, cardiovascular illness, or other systemic inflammatory diseases, the administration of cytokine therapy has been shown to induce the cytokine network, causing subsequent elevations in serum levels of IFN-γ, IL-6, IL-8, and IL-10.49,50 Furthermore, elevations in IL-6 and IL-8 correlate with symptoms of depression and anxiety.49 When exposed to a stressor, medically healthy patients with current depression exhibit resistance to molecules that normally terminate the inflammatory cascade. This suggests that patients with depression may exhibit an impaired ability to regulate inflammation.
Despite the evidence implicating inflammation in the pathophysiology of both cardiovascular disease and depression, it remains controversial whether inflammation, and cytokines in particular, is a critical etiologic link between the two. The majority of studies have evaluated small numbers of subjects and yielded equivocal results.51-55 However, the few larger studies investigating the immune response in patients with both CVD and depression were consistent.56,57 One study using population-based surveys in Germany revealed that in healthy men, a depressed mood increased the power of a concurrently elevated serum level of CRP to predict a subsequent MI.57 In this study of 3021 subjects, a baseline CRP and assessment of depressive symptoms were obtained. Three groups were formed based on the initial CRP level: low risk, medium risk, and high risk based on recent American Heart Association/Centers for Disease Control (AHA/CDC) guidelines.58 The prevalence of a depressed mood was equal among the groups. Subjects were followed for a median of 7.7 years, with a combined end point of either fatal or nonfatal MI. Patients with both high-risk levels of CRP and a depressed mood were found to have a hazard ratio of 2.69 compared with 1.55 in the group with elevated CRP alone.
In the Prospective Study of Myocardial Infarction (PRIME) study of healthy middle-aged men from France and Belfast, there was an association between depressive mood and CHD, which persisted after adjusting for inflammatory markers.56 This study compared 335 future cases of CHD to 670 matched controls. Subjects were followed for a minimum of 5 years. There was a statistically significant correlation, independent of social characteristics and classic cardiovascular risk factors, between depression scores and levels of IL-6, CRP, and intercellular adhesion molecule-1 (ICAM-1), but not fibrinogen. Furthermore, men with depressed mood had a 50% increase in the odds ratio of CHD.
Vascular endothelial dysfunction is a known risk factor for the development and progression of atherosclerosis, hypertension, and CHF. It is defined as a diminished vasoactive, anticoagulant, and anti-inflammatory status. Multiple prospective studies have confirmed that in patients with CVD, the presence of endothelial dysfunction is an independent predictor of cardiovascular events.59 Furthermore, recent studies have demonstrated a significant relationship between impaired endothelial function and both untreated and treated depression.60,61
One of the most common modalities used in the assessment of endothelial function is through measurement of brachial artery flow or flow-mediated dilation (FMD). Two small case-control studies comparing depressed patients without risk factors for CHD to age, sex, and risk-factor matched controls suggest that endothelial function, as determined by FMD, is abnormal in depressed patients.60,61 Broadley and coworkers61 evaluated patients with treated depression who were without risk factors for CHD, stable on antidepressant therapy for a minimum of 3 months, and free of depressive symptoms. This cohort showed abnormal FMD despite remission of their depressive symptoms. Sherwood and colleagues62 assessed endothelial function in patients with documented CHD; brachial artery FMD revealed a significantly higher incidence of endothelial dysfunction in patients with depressive symptomatology. The use of antidepressant medication was associated with an improved FMD, although the duration of antidepressant use was not reported. Rybakowski and coworkers63 evaluated endothelial function in controls and psychiatric patients during an acute depressive episode before and after treatment of their mood disorder. Arterial endothelial dysfunction was independent of the diagnosis (MDD vs bipolar affective disorder), intensity of depression, and type of depression, suggesting that endothelial dysfunction can be a marker of mood disorders.
Two primary components that are central to the fight or flight stress response are the HPA system and the sympathoadrenal system. In response to stress, hypothalamic neurons containing corticotropin-releasing factor (CRF) increase the synthesis and release of corticotropin (adrenocorticotropic hormone [ACTH]), β-endorphin, and other proopiomelanocortin) products from the anterior pituitary gland. Many studies have documented evidence of HPA system hyperactivity in medication-free patients with major depression: blunting of the ACTH response to CRF administration, nonsuppression of cortisol secretion after dexamethasone administration, hypercortisolemia, and pituitary and adrenal gland enlargement, and direct evidence of increased numbers of hypothalamic CRF neurons in postmortem brain tissue from depressed patients compared with controls.64 Administered corticosteroids have long been known to induce hypercholesterolemia, hypertriglyceridemia, and hypertension. Other atherosclerosis-inducing actions of steroids include injury to vascular endothelial cells and intima and the inhibition of normal healing. Indeed, elevated morning plasma cortisol concentrations have been significantly correlated with moderate to severe coronary atherosclerosis in young and middle-aged men.
Many patients with major depression also exhibit dysregulation of the sympathoadrenal system, which consists of the adrenal medulla and sympathetic nervous system (SNS). Although CNS regulation of the sympathoadrenal system has been only partially characterized, hypothalamic CRF-containing neurons provide stimulatory input to several autonomic centers that are involved in regulating sympathetic activity. Nerve impulses from regulatory centers in the CNS control catecholamine release from the sympathoadrenal system. Physiologic and pathologic conditions causing sympathoadrenal activation include physical activity, coronary artery ischemia, heart failure, and mental stress. Epinephrine in plasma is derived from the adrenal medulla, whereas plasma norepinephrine (NE) concentrations reflect the secretion of NE largely from sympathetic nerve terminals, with the remaining NE provided by the adrenal medulla and extraadrenal chromaffin cells. Peripheral plasma NE concentrations are determined not only by the rate of release from sympathetic nervous system nerve terminals, but also by reuptake into presynaptic terminals, local metabolic degradation, and redistribution into multiple physiologic compartments.
Hypersecretion of NE in unipolar depression has been documented by elevated plasma NE and NE metabolite concentrations65 and elevated urinary concentrations of NE and its metabolites,66,67 although discordant reports exist.68 Not only do depressed patients exhibit higher basal plasma concentrations of NE, those with melancholia exhibit even greater elevations in plasma NE concentrations when subjected to orthostatic challenge than do normal control subjects and depressed patients without melancholia.69 Furthermore, depressed patients who are dexamethasone (DST) nonsuppressors exhibit significantly higher basal and cold-stimulated plasma concentrations of NE than do depressed patients who are dexamethasone suppressors.69 After treatment with tricyclic antidepressants (TCAs), urinary excretion of NE and its metabolites diminish together with plasma NE concentrations,70 although Veith and colleagues65 reported that chronic treatment with desipramine increased plasma concentrations of NE. Thus sympathoadrenal hyperactivity seems to represent a state rather than a trait marker of depression, possibly reflecting increased CRF release within the CNS.
Alterations in autonomic nervous system activity, as demonstrated by exaggerated responses in heart rate to orthostatic challenge,68 reduction in heart rate variability (HRV), or abnormal cardiac vagal control,71 represent additional mechanisms of diminished survival of depressed patients with CVD. Beat-to-beat fluctuations in hemodynamic parameters reflect the dynamic response of cardiovascular control systems to a myriad of naturally occurring physiologic perturbations. Therefore, HRV can provide a sensitive measure of the functioning of the rapidly reacting sympathetic, parasympathetic, and renin-angiotensin systems.
HRV is often decreased in patients with severe CHD or heart failure.72 Moreover, the risk of sudden death after an acute MI is significantly higher in patients with decreased HRV.73 Although positive predictive accuracy is not high when HRV is considered alone, in combination with other prognostic factors, clinically useful levels of negative predictive accuracy can be achieved.74
Reduced high-frequency HRV has been observed in depressed patients in comparison with nondepressed groups,75 although discrepant reports exist.76 In patients with angiographically confirmed CHD, diminished HRV during 24-hour Holter monitoring was significantly more common in depressed patients than in matched nondepressed patients.77 Diminished high-frequency HRV is thought to reflect decreased parasympathetic tone. Diminished HRV in patients with major depression can also be secondary to a deficiency of omega-3 fatty acids.78 These polyunsaturated lipids possess antiarrhythmic properties and reduce the risk of ventricular arrhythmias.79,80 Though multiple studies have documented a deficiency of omega-3 fatty acids in patients with major depression,81 results of studies evaluating the efficacy of augmentation of selective serotonin reuptake inhibitors (SSRIs) with omega-3 fatty acids versus treatment with a SSRI alone remain equivocal.82
HRV is also reduced in patients with depression and comorbid CHD. In a comparison of 311 depressed and 367 nondepressed patients, all with recent acute MIs, patients with depression had reduced HRV and were at higher risk for all-cause mortality.83,84 Interestingly, studies have yet to show improvement of HRV after effective depression treatment, either after antidepressant or cognitive behavioral psychotherapy (CBT).85 This finding holds true for medically healthy patients or patients with comorbid CVD and may be related to the overarching changes in autonomic dysfunction, of which HRV is a parameter, as a result of depression. Heart rate recovery after exercise treadmill testing (a function of HRV) in patients with CHD is further impaired in patients with comorbid depression.86 And a recent study of 75 patients with a recurrent episode of depression compared with healthy age-matched controls showed that before treatment of the depression, there was an overall shift of autonomic balance toward sympathetic predominance.87 This shift was then exacerbated with subsequent serotonin-norepinephrine reuptake inhibitor (SNRI) or SSRI treatment, though the latter to a lesser degree.
In addition to reduced HRV, elevated nighttime heart rate (HR) has also recently been identified as a risk factor for mortality in (both depressed and nondepressed) patients after acute MI.88 It has long been established that patients with depression often experience disturbed sleep. Interestingly, depressed patients exhibit higher mean day and nighttime HR than their nondepressed counterparts, though it is the elevated nighttime HR that is predictive of mortality. It remains to be determined whether treatment aimed at addressing the cause(s) of elevated HR in this patient population would translate into improved survival.
The adverse effects of depression on cardiovascular disease have been posited to be mediated by platelet mechanisms.89 Markovitz and Matthews90 first proposed that enhanced platelet responses to psychological stress can trigger adverse coronary artery ischemic events. Musselman and coworkers,91 using fluorescence-activated flow cytometric analysis, found that in comparison with normal controls, young, medically healthy, depressed patients without any other risk factors for CHD exhibited enhanced baseline platelet activation as well as increased platelet responsiveness. Moreover, in another study, depressed patients with ≥1 traditional risk factors for CHD exhibited, under basal conditions, increased number of circulating platelets that had proceeded to irreversible degranulation.92
Indeed, patients suffering from comorbid CVD and major depression also exhibit increased platelet activation as measured by markedly elevated plasma concentrations of the platelet secretion products platelet factor 4 (PF4) and β-thromboglobulin (β-TG) compared with nondepressed, age-matched patients with CVD.93 Increased platelet activation has also been documented in CVD patients with the negative emotion, hostility, in comparison with healthy controls.94 Although the mechanism or mechanisms responsible remain unknown, heightened susceptibility to platelet activation and secretion can contribute, at least in part, to the increased vulnerability of depressed patients to CVD and/or mortality after an MI.
Serotonin secreted by platelets induces both platelet aggregation and coronary vasoconstriction, both of which are mediated by 5-hydroxytryptamine (5-HT2) receptors. Vasoconstriction occurs especially when normal endothelial cell counterregulatory mechanisms of vascular relaxation are defective, as often occurs in patients with CHD.95 Indeed, essential hypertension, elevated plasma cholesterol levels, older age, and smoking, which are well-known predisposing factors for the development of CVD, all contribute to 5-HT–mediated platelet activation. Moreover, alterations in platelet 5-HT–mediated activation have also been described in affective disorders, including major depression. Alterations in both CNS and platelet serotonergic function occur in depressed patients.96
Serotonin-mediated platelet activation can contribute to the development of atherosclerosis, thrombosis, and vasoconstriction. Even though 5-HT is a weak platelet agonist, it markedly amplifies platelet reactions to a variety of other agonists, such as adenosine diphosphate, thromboxane A2, catecholamines, and thrombin. Several investigators have reported increases in platelet 5-HT2 binding density in depressed patients.97 Moreover, the changes appear to be state-dependent in that 5-HT2 binding-site density returned to control values only in patients who showed clinical improvement. Depressed patients have been found to exhibit significant reductions in the number of platelet and brain 5-HT transporter sites. The increased 5-HT2 receptor–binding density and decreased 5-HT transporter sites suggest that depressed patients can be particularly susceptible to 5-HT–mediated platelet activation and coronary artery vasoconstriction. Decreased numbers of platelet 5-HT transporters would potentially hinder the uptake and storage of periplatelet serotonin, exposing the increased numbers of 5-HT2 receptors to 5-HT.98
Platelets from depressed patients also exhibit significantly increased elevations of intracellular free calcium concentration, [Ca2+]i, after 5-HT–induced stimulation, in comparison with controls.99 Small increases in intraplatelet calcium prime the platelet secretion and aggregation response to stimulation by 5-HT or in response to increased blood flow. Thus platelets with elevated [Ca2+]i, as are observed in depressed patients, probably would exhibit increased activation in comparison with normal comparison subjects under basal conditions or in response to shear-induced aggregation (eg, after an orthostatic challenge). More recently, antidepressants that inhibit the reuptake of serotonin into neurons (and platelets) have been shown to normalize the abnormally heightened platelet activation and secretion observed in patients with depression, without,92 and with CVD.100
The combination of a vulnerable myocardium after MI, acute ischemia, and negative emotional arousal has long been thought to trigger fatal ventricular arrhythmias. Jiang and colleagues101 followed 126 patients with CHD over a 5-year period. Mental stress-induced myocardial ischemia at baseline in CHD patients was associated with significantly higher rates of subsequent fatal and nonfatal cardiac events independently of age, baseline left ventricular ejection fraction (LVEF), and previous MI. They proposed that the relation between psychological stress and adverse cardiac events is mediated by myocardial ischemia. Although myocardial ischemia is probably the most significant factor predisposing to ventricular instability, other factors also contribute. Psychological stress in humans with CHD increases ventricular ectopic activity and increases the risk of ventricular fibrillation.102 There are several similarities between the stress response and major depression: Both can be characterized by increased blood pressure and heart rate as well as increased arousal and increased mobilization of energy stores. Particularly relevant to both the stress response and depression are the critical brain structures—the locus ceruleus and the central nucleus of the amygdala—both innervated by CRF-containing cell bodies or nerve terminals.103 The stress response and major depression differ in some respects, however. In depression, some aspects of the normal stress response escalate to a pathologic state that fails to respond appropriately to usual counterregulatory responses, resulting in a sustained version of a usually transient phenomenon (ie, hyperactivity of the HPA system or the sympathoadrenal system). Although many studies have linked stressful life events to the onset of major depression,104 some depressions are clearly endogenous (ie, they have no obvious environmental precipitant).
Frasure-Smith and colleagues proposed that depression worsens the prognosis after an MI through another mechanism: premature ventricular contractions (PVCs).21 Although the frequency of arrhythmias in depressed and nondepressed patients with CHD was similar, depressed patients with ≥10 PVCs per hour were at higher risk for sudden cardiac death than their nondepressed counterparts. Patients who were not depressed experienced little increase in risk associated with PVCs even in the presence of a low LVEF.21 Thus the prognostic impact of PVCs can be related more to depression than to PVCs per se. In the Cardiac Arrhythmia Suppression Trial (CAST),105 suppression of PVC frequency in post-MI patients did not reduce and actually increased mortality (even though PVCs are associated with increased mortality after an MI). Thus treatment of depression can be necessary to improve survival in depressed patients with PVCs.
Another electrocardiographic abnormality observed in patients with depression, as well as panic disorder, is increased QT variability. Multiple studies have shown that increased QT variability in patients with CVD is predictive of arrhythmic events and sudden cardiac death.106 Increased QT intervals have been observed in medically healthy individuals with depression107 as well as depressed patients post-MI.108 Carney and colleagues108 compared QT intervals between two groups within 28 days of an acute MI. One group’s participants met criteria for either major or minor depression, whereas the other group only included patients free of depressive symptoms. The QT interval variability was consistently higher at each of the eight sampling times over a 24-hour period in the depressed group; however, the differences were significant only at midnight and 6:00 am. Given the circadian variability of sudden cardiac death (with a peak incidence in the early morning hours), there can be a greater susceptibility to arrhythmias and sudden death in depressed, post-MI patients in the early morning.
Anxiety Disorders and Coronary Heart Disease
Anxiety disorders are the most prevalent psychiatric disorders in the United States (Tables 96–3 and 96–4), but remain largely undiagnosed and undertreated.109
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


