Effect of Noncardiac Drugs, Electricity, Poisons, and Radiation on the Heart: Introduction
Noncardiac Drugs
Chemotherapeutic agents can result in acute or chronic cardiovascular toxicity. The heart, composed of nonproliferating myocytes, was traditionally thought to be protected from the effects of drugs on rapidly dividing cells. A number of these agents are now recognized to cause cardiovascular complications including cardiomyopathy, myocarditis, pericarditis, myocardial ischemia, arrhythmias, and peripheral hypotension or vasospasm.1
Cardiovascular alterations in the patient receiving chemotherapy can be the result of a specific drug or combination of drugs or be related to tumor-associated factors such as hypercoagulability or release of myocardial depressant factors. Correlating a specific therapy with a particular adverse event can be difficult; however, knowledge of adverse effects of each agent should be considered when prescribing therapy.1-3
The anthracycline antineoplastics—doxorubicin, daunorubicin, idarubicin, and epirubicin—are the leading cause of chemotherapy-related heart disease. These agents can cause cardiac problems during therapy, weeks after completion of therapy, or, unexpectedly, years later.4 During acute therapy, electrocardiogram (ECG) changes occur in approximately 30% of patients and usually regress within weeks. Findings include ST-T wave changes, decreased QRS voltage, prolongation of the QT interval, and atrial and ventricular ectopy. Sustained atrial or ventricular arrhythmias are rare. The occurrence of early ECG abnormalities does not predict cardiomyopathy and is not an indication to discontinue therapy.1 The development of persistent sinus tachycardia in an otherwise stable oncology patient (although nonspecific), however, can raise the suspicion of ventricular dysfunction and impending congestive heart failure. Congestive heart failure is related to the cumulative dose of the anthracycline administered. The incidences of heart failure at specific doses of doxorubicin include 0.4% at 400 mg/m2 of body surface area, 7% at 550 mg/m2, and 18% at 700 mg/m2 (Fig. 94–1).5 Traditionally, the cardiac-limiting dose has been described as 550 mg/m2 because of the acute increase in heart failure seen above this dose. There is great individual variability, however, with reports of heart failure occurring with doses <100 mg/m2 and, conversely, with some patients tolerating >1000 mg/m2 without cardiac compromise.5,6 Risk factors for anthracycline-induced cardiomyopathy are debated but include prior chest radiation, young age (0-12 years of age), age >70 years, and preexisting heart disease.5-7 Young females can be at particularly increased risk for late cardiac dysfunction.7 Rapid infusion schedules associated with higher peak drug concentration appear to result in greater cardiotoxicity. Combination therapy with cyclophosphamide is an additional risk factor,1 with cardiotoxicity noted at doses of 300 mg/m2. The pathogenesis of anthracycline-induced cardiotoxicity is not known. Theories generally implicate free radical damage.8,9 The average time to clinical development of heart failure symptoms is 1 month from the end of anthracycline therapy but can occur anytime within 1 year. There is increased recognition of symptomatic heart failure occurring years after therapy. Patient presentation is similar to that for other dilated cardiomyopathies (see Chap. 32).
Figure 94–1.
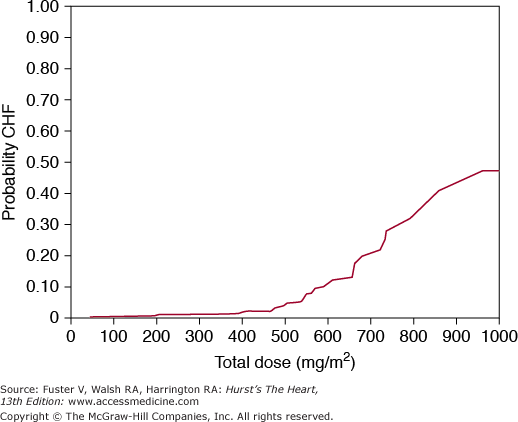
The development of doxorubicin-induced heart failure is related to cumulative dose. Toxicity may occur at any dose, but at 550 mg/m2, the probability increases significantly. CHF, congestive heart failure. Reproduced with permission from Von Hoff et al.5
Noninvasive assessment of left ventricular function has been used to guide anthracycline dosing and prevent cardiac toxicity. Serial echocardiography and/or radionuclide angiography (see Chaps. 18 and 21) are most commonly used.10,11 The most commonly used parameter is resting left ventricular ejection fraction. Adult guidelines for serial assessment have been developed. A decrease in left ventricular ejection fraction >10% (ejection fraction units) and to below a normal value of 50% is an indication to discontinue therapy. A baseline left ventricular ejection fraction <30% has generally been considered a contraindication to initiating anthracycline therapy.12-14 Unfortunately, substantial myocyte damage may occur prior to a decrease in the ejection fraction. Use of biomarkers, including troponin and B-type natriuretic peptide, to monitor toxicity has gained support but has not become clinical standard.15
There is growing recognition of the occurrence of cardiac dysfunction years after completion of anthracycline therapy. Clinical strategies for preventing anthracycline cardiotoxicity have had to balance the need for antineoplastic efficacy. Lower clinical toxicity in adults has been noted with prolonged infusions of doxorubicin over 48 to 96 hours to avoid high peak concentrations. In contrast, continuous infusion schedules in children do not offer a cardioprotective advantage.16 Due to the difficulty of implementing prolonged infusion schedules and the uncertainty of the benefits versus risks, doxorubicin continues to primarily be given by bolus infusion. Several antioxidants have been evaluated but with inconclusive results.9,17
Mitoxantrone, an anthracenedione lacking the amino sugar of anthracyclines, causes cardiotoxicity with features similar to anthracycline-induced cardiomyopathy.1 This drug appears to have less cardiotoxicity than doxorubicin at equal myelotoxic doses. Cumulative doses >160 mg/m2 are associated with an increasing incidence of congestive heart failure. There has been increasing concern that mitoxantrone in high doses and particularly when combined with other chemotherapeutic agents can result in a high incidence of delayed myocardial toxicity.18 High-dose cyclophosphamide (120-240 mg/kg over several days) used in bone marrow transplantation can cause acute cardiac toxicity.1,19 Symptomatic systolic dysfunction, usually reversible with drug discontinuation, is associated with decreased QRS voltage on the ECG. Pericardial effusions have been noted, and a hemorrhagic myocarditis can result in death. Necropsy data demonstrate endothelial injury with resultant interstitial fibrin deposition and capillary microthrombosis. The cardiotoxicity of cyclophosphamide is likely caused by damage from its biologically active metabolites. Rapid metabolizers of cyclophosphamide appear to be prone to cardiotoxicity. The metabolites cause toxic endothelial damage leading to muscle damage.19 Cyclophosphamide can also potentiate the cardiotoxic effects of the anthracyclines.1,18 Ifosfamide is an alkylating agent that can cause toxicity similar to cyclophosphamide.2
Fluorouracil can occasionally cause angina, electrocardiographic changes, and rarely myocardial infarction.1,20 The majority of episodes occur during the first cycle of therapy and resolve spontaneously after discontinuation. Arrhythmias and systolic dysfunction have been observed. The understanding of fluorouracil toxicity is complicated because combination chemotherapy is generally used, patients can be systemically ill, and many receiving this medication have preexisting coronary artery disease.20 The incidence of cardiac toxicity is uncertain but ranges from 1% to 8%.21 Patients with known coronary artery disease are at higher risk for serious cardiotoxicity. The mechanism of toxicity remains unclear, although coronary vasospasm has been suspected. Coronary catheterization has generally failed to demonstrate vasomotor hyperreactivity with fluorouracil or ergonovine challenge.
Amsacrine (AMSA P-D) has been associated with prolongation of the QT interval. Malignant ventricular arrhythmias can occur in <1% of patients and are exacerbated by hypokalemia.22
Paclitaxel (Taxol) is used to treat many types of cancer. The most common cardiovascular effect is the development of transient asymptomatic bradycardia, occurring in >10% of patients.3
Trastuzumab (recombinant humanized antihuman epidermal growth receptor 2 [HER-2] antibody) is a treatment for breast cancer that has had favorable antitumor effects when added to standard chemotherapy in selected patients.23 A retrospective review described a 27% incidence of cardiotoxicity when this agent was given with an anthracycline and cyclophosphamide, a 13% incidence in combination with paclitaxel, and a 3% to 7% incidence when given alone. The majority of these patients had received prior anthracycline therapy.24 The pathophysiology of trastuzumab cardiotoxicity is uncertain, but unlike anthracycline cardiotoxicity, ultrastructural changes have not been noted on cardiac biopsy. Clinically, symptoms occur more acutely than anthracycline toxicity, and left ventricular dysfunction is often reversible with removal of the agent.15,24,25
The tyrosine kinase inhibitors sorafenib, sunitinib, imatinib, and lapatinib have been associated with cardiotoxicity resulting in heart failure.
Interferon-α can cause supraventricular tachyarrhythmias. A reversible cardiomyopathy has been described.1,19,26,27
Psychiatric illness, particularly depression, is common in patients with cardiovascular disease (see Chap. 96). Morbidity and mortality following cardiac events are increased in patients with depression, particularly if untreated.28,29 A variety of psychotropic agents have conduction or vascular effects. A thorough understanding of these therapeutic, but potentially toxic, agents is necessary in the treatment of patients with preexisting cardiac disease. Intentional overdose with these drugs can result in serious cardiac manifestations.
The tricyclic antidepressants have several properties that account for the majority of cardiovascular effects. These drugs inhibit uptake of both norepinephrine and serotonin, resulting in greater toxicity compared with the selective serotonin reuptake inhibitors (SSRIs). A hyperadrenergic state can result in tachycardia. α-Blockade occurs at higher drug levels and can cause marked hypotension in the setting of overdose. The anticholinergic effects result in tachycardia, dry mouth, and constipation, and in overdose,30 they can delay gastrointestinal absorption of the drug. Sodium channel blockade, typical of the type IA antiarrhythmic compounds, results in conduction abnormalities and the potential to suppress ventricular function.31
The most common electrocardiographic changes include nonspecific ST-T changes and prolongation of the QT interval, PR interval, and QRS duration. PR prolongation is caused by prolonged infranodal conduction. Patients with preexisting conduction disease, particularly bundle-branch block, are at increased risk of toxicity.32 The tricyclic antidepressants have type IA antiarrhythmic properties including the potential for a proarrhythmic effect for these drugs at therapeutic doses in patients with serious structural heart disease.30 Tricyclic antidepressants are generally contraindicated in the recovery phase following myocardial infarction. Although tricyclic antidepressant therapy can be indicated in the treatment of severely depressed patients, the threshold for use should increase as the severity of heart disease increases or when there is QT prolongation.30 These issues are discussed in detail in Chap. 96.
Tricyclic antidepressants can impair left ventricular function in patients with severe systolic dysfunction; however, decreases in left ventricular ejection fraction have generally not been noted in patients with moderately impaired function.
Tricyclic antidepressant overdose carries a mortality of 2% to 3%, which is generally related to cardiac complications. Clinical status at initial presentation and serum drug levels are not predictive of prognosis. QRS prolongation is a sign of toxicity but can be absent in the patient with serious cardiac complications. Rightward deviation of the terminal 40 ms of the frontal plane QRS axis is a more sensitive marker.33 Aggressive support measures in tricyclic antidepressant overdose should be initiated immediately and include airway maintenance, gastric lavage, and repeated dosing of activated charcoal. Alkalinization with intravenous sodium bicarbonate decreases unbound drug and reverses cardiac and central nervous system conduction defects. Alkalinization is indicated in cardiac arrest, hypotension, arrhythmias, acidosis, and QRS prolongation. Hypotension refractory to volume loading and bicarbonate therapy should be treated with vasopressors, including norepinephrine or phenylephrine. Class I antiarrhythmics (eg, quinidine, procainamide, disopyramide) are contraindicated, and class III agents are potentially proarrhythmic in this setting because of prolongation of QT interval. Sodium bicarbonate is the initial therapy for ventricular dysrhythmias.34,35
SSRIs have not been studied extensively in patients with cardiac disease. Case reports of cardiac toxicity are rare, despite the increasing popularity of these agents in the treatment of depression. These agents have rarely been associated with orthostatic hypotension and bradycardia. Cardiac function does not appear to be depressed by these agents.36 The SSRIs can affect the cytochrome P450 system and can therefore alter the metabolism of a variety of drugs, including agents used in cardiovascular disease such as antiarrhythmic medications, β-blockers, calcium channel blockers, and warfarin.35-37
The monoamine oxidase (MAO) inhibitors have little effect on cardiac conduction or myocardial contractility. Orthostatic hypotension is common, particularly in elderly patients. The major concern with these agents is interaction with other drugs or tyramine-containing substances, resulting in hypertensive crisis. Lithium, used commonly in the treatment of bipolar disorder, is generally well tolerated in patients with cardiac disease. Suppression of sinus node automaticity, resulting in bradycardias, is the most common complication.38 In patients free of known heart disease, clinically significant sinus node dysfunction occurs in <1% of patients and is reversible with discontinuation of lithium therapy. Preexisting sinus node disease or concomitant therapy with drugs altering sinus node function, however, can result in sinus bradycardia. Lithium-induced hypothyroidism can be a contributing factor.39 Pacemaker therapy can be required to allow continuation of lithium therapy. Lithium therapy has been associated with electrocardiographic changes simulating hypokalemia. T wave inversion, prominent U waves, and QT prolongation can occur. PR prolongation, bundle-branch block, and complete heart block are rare.38 Overdose with lithium can result in severe bradycardias requiring temporary pacemaker therapy. A low anion gap can suggest the presence of lithium toxicity.40
The phenothiazine antipsychotic agents have potential cardiac toxicity similar to that of the tricyclic antidepressants. These drugs can cause sinus tachycardia, PR and QT prolongation, and disturbances of intraventricular conduction. Chlorpromazine and thioridazine41 are the most commonly implicated phenothiazines as causes of torsade de pointes. The butyrophenone, haloperidol, is also associated with torsade de pointes at high doses given intravenously.42
Clozapine, an atypical antipsychotic agent, has been associated with myocarditis in rare case reports.43
As discussed earlier, tricyclic, phenothiazine, and other psychotropic agents can prolong the QT interval and induce torsade de pointes. A variety of antiarrhythmic agents, particularly the type I agents, are strongly associated with this potentially fatal arrhythmia. Other toxic causes of torsade de pointes44 are listed in Table 94–1.
| Drugs commonly involved |
| Dofetilide |
| Ibutilide |
| Procainamide |
| Quinidine |
| Sotalol |
| Bepridil |
| Other drugs (<1% incidence) |
| Amiodarone |
| Arsenic trioxide |
| Cisapride |
| Anti-infective agents: clarithromycin, erythromycin, halofantrine, pentamidine, sparfloxacin |
| Antiemetic agents: domperidone, droperidol |
| Antipsychotic agents: chlorpromazine, haloperidol, mesoridazine, thioridazine, pimozide |
| Methadone |
The QT prolongation and torsade de pointes reported with the antihistamines terfenadine and astemizole and with cisapride have been associated with high drug levels from excessive dosing or altered metabolism.45,46 These drugs have been removed from the market in the United States. Terfenadine-, astemizole-, and cisapride-induced prolongation of the QT interval is caused by the electrophysiologic activity of blocking human ether-a-go-go-related gene (HERG), the ion channel that is responsible for the rapid component of the delayed rectifier current for potassium (IKr).46 These drugs are metabolized by cytochrome P450 3A. A variety of agents inhibit this isoenzyme, including antifungals (eg, ketoconazole, fluconazole, itraconazole), erythromycin or clarithromycin (not azithromycin), SSRIs (eg, fluvoxamine, nefazodone, fluoxetine, sertraline), quinine, and grapefruit juice. Serious cardiac arrhythmias have been reported in patients taking terfenadine, astemizole, or cisapride with drugs that inhibit the cytochrome P450 3A isoenzyme.
Valvular heart disease, resembling that seen with carcinoid syndrome, has been associated with the antimigraine drugs methysergide and ergotamine, with the weight loss medications dexfenfluramine and fenfluramine, and, in several instances, with pergolide mesylate used to treat Parkinson disease and restless leg syndrome.47-55 The incidence of valvular abnormalities is more common with methysergide compared with ergotamine and appears to be greater with the chronic use of dexfenfluramine and the combination of fenfluramine and phentermine. Dexfenfluramine and fenfluramine were withdrawn from the market in 1997 when up to 30% of users were reported to develop asymptomatic valve regurgitation.50-53
In addition to ergotamine and methysergide, sumatriptan is used to treat migraines. Sumatriptan, a selective serotonin type I agonist, can cause coronary artery vasospasm. Sumatriptan should not be taken within 24 hours of treatment with ergotamine-like medications because of the risk of prolonged vasoconstriction.56
Ergotamine, methysergide, and sumatriptan are generally contraindicated in patients with obstructive coronary artery disease because of vasoconstrictor effects and the possibility of precipitating angina.57
The antimalarial agent chloroquine is commonly used to treat collagen vascular and dermatologic disorders. Irreversible retinal damage is the primary concern with long-term or high-dose therapy. Skeletal myopathy and, less commonly, cardiomyopathy can occur. With cardiac involvement, features of restrictive cardiomyopathy are most common. Myocardial biopsy with analysis by electron microscopy showing curvilinear and myeloid bodies is diagnostic. These findings can be seen on skeletal muscle biopsy. The ECG can demonstrate T wave changes and conduction abnormalities. Acute chloroquine poisoning results in hypotension, tachycardia, and prolongation of the QRS and is often fatal.58,59
Illicit use of androgens has been identified as a problem in competitive athletes and body builders. It is estimated that 300,000 persons in the United States have had recent steroid use, and >1 million have had prior use.60-62 Anabolic steroids, including testosterone, stanozolol, and nandrolone, are frequently used in combination and at high doses for intermittent periods of several weeks to months. Doses commonly exceed 100 times the doses used for medical purposes.60 Animal data indicate that these agents can cause abnormal lipids, left ventricular hypertrophy, increased blood volume, and hypertension. Data on human toxicity related to vascular or myocardial abnormalities are inconclusive.63 Stanozolol and nandrolone reduce total high-density lipoprotein levels by >50% and increase low-density lipoprotein levels by >30%.64 Isolated reports of young men (<35 years of age) developing severe coronary atherosclerosis, myocardial infarctions, or stroke exist in the literature.60,65 Because of the secrecy surrounding the use of these agents, the full clinical significance of abuse is not known.
Cocaine has a generalized sympathomimetic effect and has local anesthetic properties. Cocaine blocks the reuptake of norepinephrine and dopamine on preganglionic sympathetic nerve terminals. This produces sympathetic stimulation both centrally and peripherally. These catecholamine effects acutely result in tachycardia, hypertension, increased myocardial contractility, and vascular constriction. The local anesthetic effect, occurring through blockade of the fast sodium channel, results in slowed conduction in myocardial tissues. This can result in electrocardiographic abnormalities including prolongation of the PR, QRS, and QT intervals similar to that seen with toxicity from type I antiarrhythmic agents. These effects increase the vulnerability to reentrant ventricular arrhythmias.66-68
Cocaine can result in increased thrombogenicity.69 Platelet aggregations is enhanced, and endothelial function is altered, resulting in the potential for development of coronary thrombosis in the absence of coronary atherosclerosis.70 Chronic use of cocaine is associated with premature coronary atherosclerosis.71 Cocaine indirectly causes constriction of both diseased and nondiseased coronary artery segments, but its effect is more marked in diseased vessels. Up to one-third of reported cases of patients with cocaine-induced myocardial infarctions have normal coronary arteries.67 The combined cardiac effects, including early coronary atherosclerosis, coronary vasospasm, increased thrombogenicity, increased myocardial oxygen demands, and proarrhythmic effects, make this drug a lethal threat to users of all ages.
Chest pain is the most common reason for cocaine users to seek medical attention. More than 64,000 patients are evaluated annually for cocaine-related chest pain, of whom more than one-half are admitted to the hospital. The evaluation of cocaine-related chest pain is difficult.72,73 Prospective studies demonstrate that approximately 6% of patients presenting to the emergency room with cocaine-related chest pain have myocardial infarction. These patients are often young men without other risk factors for coronary artery disease except for tobacco smoking. The duration and quality of discomfort does not readily distinguish those eventually noted to have enzyme documentation of infarction. Many young patients have early repolarization patterns, with ST elevation in leads V1 to V3, a normal variant that can be confused with acute infarction. Infarction has been noted in patients with normal or nonspecific ECGs. Because of the difficulty in excluding myocardial infarctions, patients are often monitored for a period of at least 12 hours until enzymes have excluded infarction.72
Treatment strategies for cocaine-induced myocardial ischemia have been developed based on the known cardiac and nervous system toxicity of the drug.66,70
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


