CHAPTER 130 Ebstein’s Anomaly
HISTORICAL ASPECTS
In 1866, Dr. Wilhelm Ebstein, a young physician in Breslau, Poland, described the unusual cardiac findings of a 19-year-old laborer who had died of cyanotic heart disease.1 In addition to describing the characteristic anatomic findings in this anomaly, Dr. Ebstein accurately described the hemodynamic abnormalities and correlated them with the patient’s signs and symptoms. During his lifetime, Dr. Ebstein was praised for other contributions he made to pathology and medicine, but his students and colleagues overlooked his classic description of the congenital cardiac anomaly with which his name is identified today.2
EMBRYOLOGY AND PATHOLOGIC ANATOMY
The exact embryology of Ebstein’s anomaly is unknown. The leaflets and the tensor apparatus of the tricuspid valve are thought to be formed at a relatively late stage in cardiac development by a process of delamination of the inner layers of the inlet portion of the right ventricle.3,4 The insertions of the septal and posterior leaflets (and occasionally the right lateral aspect of the anterior leaflet) are displaced to the junction of the inlet and trabecular components of the right ventricle, suggesting failure of delamination of these leaflets.5
The atrialized ventricle is characteristically thinned and dilated, but careful observation shows that the entire wall of the right ventricle, both proximal and distal to the abnormal insertion of the tricuspid leaflets, including the infundibulum, is also dilated. Dilation of the right ventricular wall is associated not only with thinning of the wall but also with an absolute decrease in the number of myocardial fibers.6 The atrioventricular node is located at the apex of the triangle of Koch, and the conduction system is in its normal position. Atrial septal defect and other associated anomalies are common.
In those congenital cardiac anomalies in which there is situs solitus and atrioventricular discordance with ventriculoarterial discordance (corrected transposition), Ebstein’s anomaly of the left-sided (systemic, morphologically tricuspid) atrioventricular valve may be found. The nature of the displacement of the septal and posterior leaflets in left-sided Ebstein’s anomaly is similar to that in the right-sided form, but the anterior leaflet is smaller and anatomically different.7 Other differences relate to the functional portion of the morphologically right ventricle, which is rarely dilated in young patients, and the atrialized portion of the ventricle, which has less thinning of the wall. The atrioventricular conduction tissue in corrected transposition is right sided and anterior, at a distance from the left-sided tricuspid valve.8 The remainder of this chapter focuses on the classic right-sided Ebstein’s anomaly.
CLASSIFICATIONS OF EBSTEIN’S ANOMALY
Carpentier’s classification was proposed in 1988 to describe four types of Ebstein’s anomaly based on morphology of the right ventricle and tricuspid valve (Table 130-1).9 An alternative classification, proposed in the Congenital Heart Surgery Nomenclature and Database Project,10 was based on the feasibility of tricuspid valve reparability. In the Carpentier classification, type I occurs when the anterior leaflet is large and mobile with complete delamination of 50% or more of its surface area. The leading edge is variable. The posterior and septal leaflets may be present or absent, dysplastic, fenestrated, or displaced. The atrialized right ventricular size can vary. In type II, the anterior, posterior, and septal leaflets are present, small, and displaced toward the apex. There is some mobility of the valve leaflets, and often the leading edge of the anterior leaflet is freely mobile. The atrialized right ventricle chamber is moderately large. In type III, the anterior leaflet has restricted motion, with shortened, fused, and tethered cords, and it may have direct insertion of papillary muscles. The posterior and septal leaflets are displaced and dysplastic. The atrialized right ventricle is large. In type IV, the anterior leaflet is severely deformed and displaced into the right ventricular outflow tract. There may be few or no cords, and direct insertions of papillary muscles into the leading edge of the valve are common. The posterior leaflet is dysplastic or absent, and the septal leaflet is represented by a ridge of fibrous tissue descending apically from the membranous septum. Tricuspid valve tissue displaced into the right ventricular outflow tract may cause obstruction of blood flow (functional pulmonary stenosis). Almost the entire right ventricular ventricle is atrialized.
Table 130–1 Carpentier’s Classification of Ebstein’s Anomaly
| Type | Right Ventricle | Tricuspid Valve |
|---|---|---|
| A | ||
| B | ||
| C | ||
| D | Almost completely noncontractile atrialized right ventricle (except for infundibulum) |
PHYSIOLOGY
As a consequence of atrial dilation, atrial tachyarrhythmias are common and become more likely as time goes on. In addition, approximately 15% of patients will have one or more accessory conduction pathways associated with Wolff-Parkinson-White syndrome, and 1% to 2% of patients will have atrioventricular nodal reentrant tachycardia (AVNRT).11,12 In end-stage heart failure, ventricular arrhythmias can be present.
Fetuses
Presentation and Diagnosis
Ebstein’s anomaly can be diagnosed in utero using fetal echocardiography in as early as the 18th week of pregnancy.13,14 Maternal lithium use may be a risk factor for the development of Ebstein’s anomaly in the fetus,15 and there are a few reports of familial Ebstein’s anomaly.16–18 The first presentation of Ebstein’s anomaly in the fetus may include cardiomegaly, right atrial enlargement, tricuspid valve regurgitation, arrhythmia, or hydrops. Fetal echocardiography remains a challenge because of the small size of the heart, and it may be difficult to distinguish Ebstein’s anomaly from pulmonary atresia or other causes of tricuspid regurgitation.
Natural History
The prognosis of Ebstein’s anomaly in the fetus is very poor. In a study of fetuses with Ebstein’s anomaly or tricuspid valve dysplasia from Children’s Hospital Boston, eight (24% of 33) pregnancies were terminated, nine (27%) fetuses died in utero, and 16 (49%) fetuses survived to birth. Only seven (21% of 33) prenatally diagnosed patients survived beyond the neonatal period.19 Survival was related to the severity of Ebstein’s anomaly and the presence of right or left ventricular function. Elective early delivery may be needed in those fetuses who exhibit signs of distress.
Neonates and Infants
Presentation and Diagnosis
In the early neonatal period, any degree of tricuspid regurgitation is accentuated by the normally occurring elevated pulmonary arteriolar resistance, and infants with Ebstein’s anomaly may develop severe heart failure. Because the foramen ovale is often patent in early infancy, severe tricuspid regurgitation, with its resultant elevation of right atrial pressure, will produce a right-to-left atrial-level shunt, and afflicted infants may be deeply cyanotic. If the infant survives this critical period, the degree of cyanosis and the symptoms often diminish as fetal pulmonary hypertension regresses.
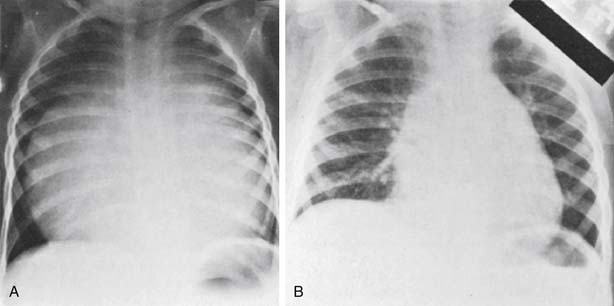
On the chest radiograph, profound cardiomegaly may be evident (Fig. 130-1). Echocardiography is the mainstay of diagnosis. Early after birth, decreasing pulmonary vascular resistance with pulmonary vasodilators may be helpful to unload the right ventricle and improve ventricular function. In some cases, a large patent ductus arteriosus may allow a circular shunt, in which blood flows from the aorta, to the patent ductus arteriosus, to the right ventricle, to the right atrium, to the left atrium, and then to the left ventricle.20 If this occurs, treatment should include cessation of prostaglandins, with resultant closure of the ductus.
Natural History
When the diagnosis of Ebstein’s anomaly is made in infancy, the prognosis is also poor. Survival for patients diagnosed between birth and 2 years of age was only 68% in one series.21 Important features that can be determined echocardiographically and that predict outcome in neonates with Ebstein’s anomaly include the patency of the right ventricular outflow tract, and the Great Ormond Street echocardiography (GOSE) score.14 The GOSE score, as described by Celermajer, is calculated in the four-chamber view to create a ratio of the combined areas of the right atrium and atrialized right ventricle compared with the functional right ventricle, left atrium, and the left ventricular areas. Patients who have the most severe GOSE score (grades 3 and 4) have a very poor prognosis (Table 130-2).14,22–24
Table 130–2 Celermajer Index and the Estimated Risk of Mortality
| GOSE Score | Index (RA + aRV): (RV + LA + LV) | Risk of Mortality∗ (%) |
|---|---|---|
| 1 | <0.5 | 0 |
| 2 | 0.5-0.99 | 10 |
| 3 | 1-1.49 | 44-100 |
| 4 | ≥1.50 | 100 |
aRV, atrialized right ventricle; GOSE, Great Ormond Street echocardiography; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
∗ From references 14, 22, and 23.
Modified from Jaquiss RDB, Imamura M. Semin Thorac Cardiovasc Surg 2007;19:258-63.
Operative Management
One approach to repair of neonatal or infant Ebstein’s anomaly is the biventricular strategy, popularized by Knott-Craig and coworkers.25,26 In this method, the tricuspid valve is repaired and the atrial septal defect is partially closed. Multiple methods have been described to improve the chance of successful repair, but they depend on having an anterior leaflet that can be mobilized; the repair is generally a monocusp repair based on a satisfactory anterior leaflet.27 The partial closure of the atrial septal defect allows right-to-left shunting, which may be necessary in the early postoperative period when there is high risk of right ventricular dysfunction and elevated pulmonary vascular resistance. Right atrial reduction is performed routinely and is important to reduce the size of the markedly enlarged heart to allow room for the lungs.
Although early mortality for this operation is high (about 25%), the intermediate outcome with the biventricular approach appears to be promising. In 2007, Knott-Craig and colleagues published their experience with 27 neonates and young infants.26 These patients had concomitant anatomic or functional pulmonary atresia (n = 18), ventricular septal defect (n = 3), small left ventricle (n = 3), and hypoplastic branch pulmonary arteries (n = 3). Twenty-five patients received a biventricular repair with tricuspid valve repair in 23, and two received a valve replacement. Survival to hospital dismissal was 74%, and there were no late deaths (median follow-up, 5.4 years; maximum, 12 years). All patients were in functional New York Heart Association (NYHA) class I. Although these early results for repair of Ebstein’s anomaly during the neonatal period are poor compared with many other neonatal anomalies corrected in the first month of life (e.g., arterial switch procedure, Norwood stage I), they have become a benchmark for this very difficult patient population.
Alternatively, the right ventricular exclusion approach, pioneered by Starnes and colleagues, involves patch closure of the tricuspid valve orifice, enlarging the interatrial connection, and placing a systemic-to-pulmonary artery shunt.28 This approach is particularly appealing in patients who also have anatomic right ventricular outflow tract obstruction. A small fenestration (using a 4- to 5-mm punch) is placed in the tricuspid valve patch23,29,30 to allow right ventricular decompression as it passively fills with blood from the thebesian veins. This also allows progressive involution of the enlarged dysfunctional right ventricle, which is helpful in the long term when preparing for the eventual Fontan procedure. In patients who have a patent right ventricular outflow tract, a competent pulmonary valve is required to prevent blood from entering the right ventricle, resulting in distention. If patients have an incompetent pulmonary valve, the main pulmonary artery should be ligated. These issues must be considered because significant dilation of a poorly functioning right ventricle eventually results in impairment of left ventricular function. Similar to the biventricular approach, right atrial reduction is routinely performed to allow space for the lungs.
A modification to the Starnes single-ventricle approach, total right ventricular exclusion, has been advocated by Sano and associates, in which the free wall of the right ventricle is resected and closed primarily or with a polytetrafluoroethylene patch.31 This procedure acts like a large right ventricular plication. This adaptation of the Starnes method may improve left ventricular filling and provide decompression to the lung and left ventricle.
Results of the single-ventricle pathway have been reported by Reemtsen and colleagues.23 Of 16 neonates, two patients had tricuspid valve repair, one patient had heart transplant, 10 patients had a right ventricular exclusion procedure with a fenestrated tricuspid valve patch, and three patients had a right ventricular exclusion procedure with a nonfenestrated tricuspid valve patch. The operative survival was 80% (8 of 10) in patients with a fenestration, and 33% in patients with no fenestration, leading the authors to recommend fenestration of the tricuspid valve patch. Among the nine hospital survivors of right ventricular exclusion, three underwent completion of the Fontan, and all nine have had a successful bidirectional cavopulmonary shunt (second stage).
Heart transplantation remains an option in the most severe cases of Ebstein’s anomaly, but it is rarely necessary in the current era because of the improved results with the Knott-Craig and Starnes approaches. Limitations of heart transplantation include the scarcity of donor organs in the neonatal age group and the side effects of long-term immunosuppression and its associated complications in the transplant recipient. Finally, the development of smaller ventricular assist devices and advances in extracorporeal membrane oxygenation have provided mechanical support options in the perioperative period for these infants.32–34
Children and Adults
Presentation and Diagnosis
The electrocardiogram is usually abnormal, but it is not diagnostic. Complete or incomplete right bundle branch block and right-axis deviation are typically present, and atrial arrhythmias are common. On chest radiograph, the cardiac silhouette may vary from almost normal to the typical Ebstein’s configuration, which consists of a globular-shaped heart with a narrow waist similar to that seen with pericardial effusion. This appearance is produced by enlargement of the right atrium and displacement of the right ventricular outflow tract outward and upward. Vascularity of the pulmonary fields is either normal or decreased.
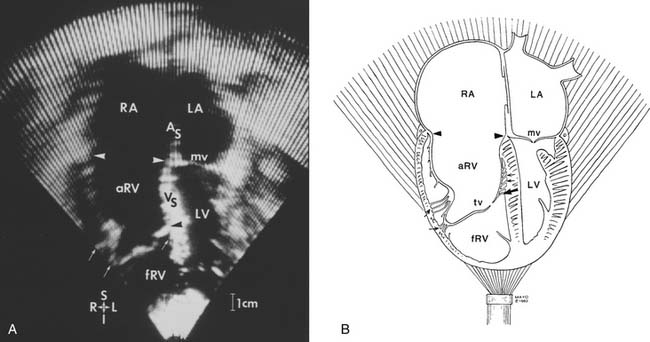
Echocardiography allows an accurate evaluation of the tricuspid leaflets and subvalvular apparatus (displacement, tethering, dysplasia, absence), the size of the right atrium, size and function of the atrialized portion of the right ventricle, and the size and function of the right and left ventricles. Doppler echocardiography and color-flow imaging allow detection of an atrial septal defect and the direction of shunt flow. The principal echocardiographic characteristic that differentiates Ebstein’s anomaly from other forms of congenital tricuspid regurgitation is the degree of apical displacement of the septal leaflet at the crux of the heart (≥0.8 cm/m2).35 In addition, color-flow imaging allows assessment of the site and degree of tricuspid valve regurgitation (Fig. 130-2). Factors that are favorable for valve repair include a large, mobile anterior leaflet with a free leading edge. Significant leaflet tethering (i.e., adherence of the edge or body of the leaflet to underlying endocardium) and the presence of tricuspid leaflet tissue in the right ventricular outflow tract make successful valve repair more difficult. Any delamination of inferior leaflet tissue is helpful, and the more septal leaflet tissue present, the more likely a successful repair can be obtained.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


