Duchenne and Becker muscular dystrophies are caused by mutations in dystrophin. Cardiac manifestations vary broadly, making prognosis difficult. Current dystrophin genotype–cardiac phenotype correlations are limited. For skeletal muscle, the reading-frame rule suggests in-frame mutations tend to yield milder phenotypes. We performed dystrophin genotype–cardiac phenotype correlations using a protein-effect model and cardiac magnetic resonance imaging. A translational model was applied to patient-specific deletion, indel, and nonsense mutations to predict exons and protein domains present within truncated dystrophin protein. Patients were dichotomized into predicted present and predicted absent groups for exons and protein domains of interest. Development of myocardial fibrosis (represented by late gadolinium enhancement [LGE]) and depressed left ventricular ejection fraction (LVEF) were compared. Patients (n = 274) with predicted present cysteine-rich domain (CRD), C -terminal domain (CTD), and both the N -terminal actin-binding and cysteine-rich domains (ABD1 + CRD) had a decreased risk of LGE and trended toward greater freedom from LGE. Patients with predicted present CTD (exactly the same as those with in-frame mutations) and ABD1 + CRD trended toward decreased risk of and greater freedom from depressed LVEF. In conclusion, genotypes previously implicated in altering the dystrophinopathic cardiac phenotype were not significantly related to LGE and depressed LVEF. Patients with predicted present CRD, CTD/in-frame mutations, and ABD1 + CRD trended toward milder cardiac phenotypes, suggesting that the reading-frame rule may be applicable to the cardiac phenotype. Genotype–phenotype correlations may help predict the cardiac phenotype for dystrophinopathic patients and guide future therapies.
Mutations in dystrophin ( DMD gene) cause Duchenne and Becker muscular dystrophies (DMD and BMD, respectively). The onset and progression of cardiac involvement are quite variable in DMD and/or BMD, making prognosis and therapy difficult. However, differences in dystrophin genotypes may explain some of the cardiac phenotype variability. Dystrophin genotype– skeletal muscle phenotype correlations suggest that in-frame mutations resulting in a semifunctional dystrophin protein result in less-severe skeletal muscle phenotype (BMD) than frameshift or early truncating mutations resulting in nonfunctional dystrophin (DMD). Previous investigations of dystrophin genotype– cardiac phenotype correlations have found dystrophin exons 45, 48 to 49, and 51 to 52, and the N -terminal actin-binding domain (ABD1), rod, hinge-III, cysteine-rich domain (CRD), and C -terminal domains (CTD) correlated with earlier onset of cardiac dysfunction. These studies focused on mutation type and location without regard to the predicted dystrophin structure and used age of onset of depressed left ventricular ejection fraction (LVEF) by echocardiogram. We aimed to perform a large-scale dystrophin genotype–cardiac phenotype correlation study using a novel direct translation model with phenotyping by cardiac magnetic resonance imaging of myocardial performance and fibrosis as seen on late gadolinium enhancement (LGE) imaging.
Methods
All boys with genetically confirmed DMD or BMD who underwent clinical CMR studies at Cincinnati Children’s Hospital Medical Center (CCHMC) from January 2005 and January 2013 were included. For the evaluation of dystrophin genotype–cardiac phenotype correlations, we included only patients with whole-exon deletion, indel, and nonsense mutations known or predicted to be disease causing (described in the following). The institutional review board approved the study. For each patient with DMD and/or BMD who had undergone a CMR study, we reviewed clinically obtained dystrophin mutation analysis. Several clinical diagnostic laboratories were used during the study period, and methods used for molecular analysis included Southern blot, polymerase chain reaction, single condition amplification/internal primer, comparative genomic hybridization, and/or multiplex ligation-dependent probe amplification. For classification of the mutation data for the cohort, each clinical diagnostic test result was checked using the Leiden reading-frame tool and then analyzed against the Leiden whole exon change database, the Leiden point mutation database, and/or the Universal Mutation Database as appropriate for the specific mutation. Mutations previously described as disease causing, or mutations expected to change the coding sequence of dystrophin, were considered pathogenic. Mutations were defined as nonsense if a base pair change created a premature stop codon at the mutation site, indel if there was an insertion or deletion of 1 to 4 nucleotides that resulted in a premature stop codon, splicing if they occurred at a predicted splice site as reported by the diagnostic laboratory, and intronic if they occurred outside both the coding and splice site sequences, as defined by the diagnostic laboratory.
To predict the presence or absence of critical functional protein domains, we first determined which base pairs were predicted to be present for each patient based on their specific mutation. A direct translation model was then used for whole-exon deletion, indel, and nonsense mutation types. These mutational mechanisms are predicted to result in a truncated protein and/or protein missing specific domains. The model assumed that the mRNA resulting from the patient-specific mutations was stable and not subject to nonsense-mediated decay. The predicted mRNA was then translated to determine the predicted presence or absence of each critical functional protein domain. Exon boundaries were extracted from GenBank (accession NM_004006.2 ), and protein domain boundaries were extracted from the eDystrophin project and GenBank (accession NM_004006.2 ). For out-of-frame whole-exon deletion mutations, we predicted that exons and protein domains encoded entirely 5′ to the deletion start site would be present. For in-frame whole-exon deletion mutations, we predicted that exons and protein domains coded entirely either 5′ or 3′ of the deleted segment would be present. For indel and nonsense mutations, we predicted that exons and protein domains encoded entirely 5′ to the mutation site would be present. The creation of new 3′ protein domains in patients with out-of-frame mutations was not incorporated into the model. For each patient, we then determined whether each exon and protein domain of interest was predicted to be present or absent.
Image acquisition for our DMD/BMD cohort has been described previously. The CMR studies were conducted on a clinical 3- or 1.5-T scanner, depending solely on schedule availability. At CCHMC, we routinely perform CMR on every DMD and BMD patient annually; only patients who refused or could not tolerate lying in the scanner did not undergo the study, and an annual CMR study was recommended regardless of previous refusal or inability to undergo CMR. No anesthesia or sedation was used for these studies. The LVEF was assessed using standard planimetry techniques (QMASS MR, version 7.5; Medis Medical Imaging Systems, Leiden, Netherlands) by an expert reader (RJF, KNH, JJS, and MDT). The LVEF was defined as depressed if it was <55%. Our interobserver variability was ∼4% and intraobserver variability was ∼2% for LVEF (unpublished data). LGE was considered positive if any left ventricular segment showed subepicardial or midmyocardial hyperenhancement by visual inspection.
For each region of interest, we compared patients in the predicted present group against those in the predicted absent group. For overall risk of development of LGE and depressed LVEF, we used chi-square analyses; for age of development of LGE and depressed LVEF, we used t tests; and for freedom from LGE and depressed LVEF, we used Kaplan–Meier log-rank analyses (SAS version 9.3, SAS Institute, Cary, North Carolina). All tests were 2-sided, and a p value of <0.05 was considered statistically significant.
Results
The dystrophinopathic cohort contained 322 patients for whom genotype data were available. The mutation distribution was similar to other reported populations : 212 (66%) whole-exon deletions (30 [9.3%] in-frame and 182 [57%] out-of-frame); 39 (12%) whole exon duplications; 39 (12%) nonsense mutations; 23 (7.1%) indel mutations (19 [5.9%] deletion and 4 [1.2%] insertion); 7 (2.2%) splicing mutations; and 2 (0.6%) intronic mutations. Patients ranged in age from 4.9 to 29.7 years (mean 12.3, median 11.4 years) at time of CMR.
We analyzed the dystrophin mutations and cardiac phenotype of patients who met the inclusion criteria for genotype analysis. This subset of patients comprised 274 patients, who ranged in age from 4.9 to 29.4 years (mean 12.2 ± 4.0, median 11.3 years). Of the 274 patients, 11 (4.0%) were patients with BMD. During the study, 15 patients (5.5%) died. Of the 274 in the study, 231 (84%) had been treated with steroids: 138 (50%) with deflazacort only, 38 (14%) with prednisone only, and 55 (20%) with both. In terms of their CMR findings, 118 patients (43%) had at least 1 LGE-positive study; 40 (15%) had at least 1 study with depressed LVEF; 32 (12%) had at least 1 study with both LGE and depressed LVEF; 126 (46%) had 1 study with either LGE or depressed LVEF, and 148 (54%) had all studies with neither LGE nor depressed LVEF. Of the 766 CMR studies examined, 183 (24%) were LGE positive, 539 (70%) were LGE negative, and 44 (5.7%) were LGE indeterminate; 692 (90%) had a normal LVEF; and 74 (10%) had depressed LVEF.
Patients were dichotomized to either the predicted present or predicted absent group for each region of interest based on the process detailed previously. The subset of patients with in-frame deletions overlapped exactly with patients predicted to have the CTD present ( Figure 1 ).
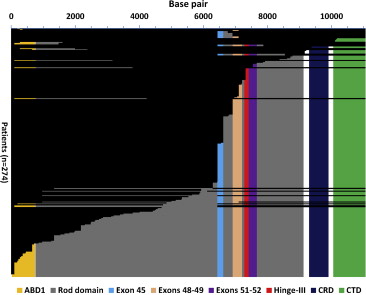
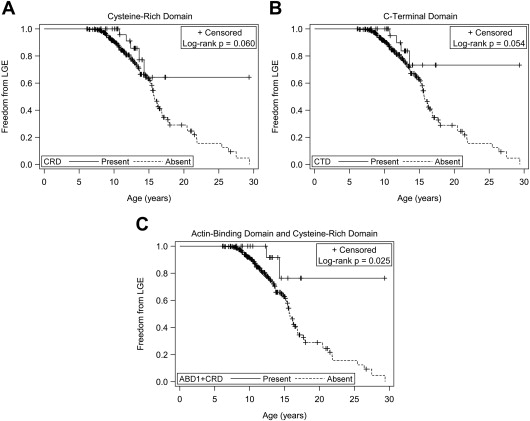
Patients predicted to have exons 45, 48 to 49, 51 to 52, or 1 to 45 present did not show a significant difference in the risk of, age of onset of, or freedom from LGE compared with the respective predicted absent groups. Patients predicted to have the ABD1, hinge-III region, or rod domains present also did not show a significant difference in risk of, age of onset of, or freedom from LGE compared with the respective predicted absent groups. Patients predicted to have the CRD and CTD present did show a decreased risk of LGE (relative risk [RR] 0.40 and 0.37, respectively, Table 1 ), but there was no significant difference in age of onset of LGE compared with the predicted absent groups. There was a trend toward greater freedom from LGE for patients with predicted present CRD (25% time-to-event 14.3 vs 13.0 years, Figure 2 ) and CTD (25% time-to-event 13.6 vs 13.0 years, Figure 2 ). We then considered patients who were predicted to have both the ABD1 and CRD present (ABD1 + CRD). Patients predicted to have both ABD1 + CRD present had a lower risk of developing LGE (RR 0.27, Table 1 ) and greater freedom from LGE ( Figure 2 ) compared with those without both predicted to be present. There was no significant difference in age of onset of LGE.
| Exon/domain (n=274) | Predicted intact (%) | Decreased risk of LGE | Freedom from LGE | Decreased risk of depressed LVEF | Freedom from depressed LVEF |
|---|---|---|---|---|---|
| CRD | 34 (12%) | 0.012 | 0.060 | 0.191 | 0.194 |
| CTD/in-frame deletions | 30 (11%) | 0.012 | 0.054 | 0.095 | 0.090 |
| ABD1+CRD | 21 (7.7%) | 0.014 | 0.025 | 0.331 | 0.132 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


