Diagnosis and Management of Heart Failure: Introduction
In his classic 1933 text, Diseases of the Heart,1 Sir Thomas Lewis identified the diagnosis and management of chronic heart failure as the cardinal problem in clinical cardiology. This observation is relevant today, as heart failure represents one of the most rapidly growing and costly forms of cardiovascular disease. As discussed in Chap. 27 both the incidence and prevalence of heart failure are substantial and rising, because heart failure remains a principal complication of virtually every form of heart disease. Moreover, heart failure is associated with high rates of morbidity, mortality, and economic cost. For example, it is estimated that at any time 30% to 40% of heart failure patients are determined to be in New York Heart Association (NYHA) functional class III or class IV, indicating an advanced degree of disability.2 Readmission rates for heart failure remain high, and 5-year mortality ranges from 15% for those with asymptomatic disease to more than 50% in patients with advanced heart failure.3-6 Fortunately, a sound understanding of the pathophysiology of the disease (see Chap. 26) and a systematic approach to heart failure evaluation and management as reviewed in this chapter results in improved patient outcomes.
In general, the current evaluation and management of patients with chronic systolic heart failure has been well-studied. Recommendations for its treatment are supported by numerous randomized controlled trials or by substantial clinical/observational experience. This evidence base has led to the publication and update of national and international guidelines directing the evaluation and management of chronic systolic heart failure in adults.7-10 In contrast, the treatment of diastolic heart failure remains largely empirical and is directed toward controlling symptoms by reducing ventricular filling pressures without reducing cardiac output. Likewise, the treatment of acutely decompensated heart failure has been inadequately studied. While published guidelines address the management of decompensated heart failure, recommendations are generally based on consensus expert opinion rather than randomized controlled trials.7-11
General Principals of Management
Preferably, heart failure should be prevented through the early treatment of risk factors and, when present, asymptomatic left ventricular dysfunction. The first revision to the 1995 American College of Cardiology/American Heart Association Guideline for the Evaluation and Management of Heart Failure developed a framework for heart failure prevention.12 The guideline, published in November 200112 and updated in September 2005,7 views heart failure as a continuum beginning with risk factors and culminating in end-stage or refractory disease. According to the guideline, there are known risk factors and structural prerequisites leading to the development of left ventricular systolic and/or diastolic dysfunction and the clinical syndrome of heart failure.
The guideline outlines four stages describing the progression of heart failure (Table 28–1).7Stage A describes those patients who exhibit one or more risk factors for the development of heart failure. If inadequately treated, the risk factors, such as hypertension, diabetes, and coronary artery disease frequently lead to the development of a structural abnormality of the heart. Stage B is defined by the development of such a structural abnormality of the heart but no symptoms of heart failure. This is the true asymptomatic or “never been symptomatic“ stage of cardiovascular disease progression to heart failure. Examples of progression from stage A to stage B include the development of left ventricular hypertrophy in the hypertensive subject or the onset of a left ventricular wall motion abnormality and reduced ejection fraction in the coronary artery disease patient following myocardial infarction. Stage C is heralded by the onset of heart failure symptoms, including shortness of breath, fatigue, and exercise intolerance. By definition, stage C patients have current or prior symptoms of heart failure. Thus, even if made asymptomatic with treatment, stage C patients are not considered to have regressed to stage B. Finally, stage D defines the patient with marked heart failure symptoms at rest despite maximal medical therapy. These patients are generally seen as being in need of heroic measures, such as cardiac transplantation or mechanical assistance, or referral into a program focused on end-of-life care.
| Stage | Definition | Patient Description |
|---|---|---|
| A | High risk for developing heart failure (HF) | Hypertension |
| Coronary artery disease | ||
| Diabetes mellitus | ||
| Family history of cardiomyopathy | ||
| B | Asymptomatic HF | Previous myocardial infarction (MI) |
| LV hypertrophy or systolic dysfunction | ||
| Asymptomatic valvular disease | ||
| C | Symptomatic HF | Known structural heart disease |
| Shortness of breath and fatigue | ||
| Reduced exercise tolerance | ||
| D | Refractory end-stage HF | Marked symptoms at rest despite maximal medical therapy (eg, those who are recurrently hospitalized or cannot be safely discharged from the hospital without specialized interventions |
The treatment of patients with risk factors (stage A) and asymptomatic left ventricular dysfunction (stage B) is extensively reviewed elsewhere in this book and briefly in this chapter. Once left ventricular dysfunction and symptomatic heart failure ensue (stages C and D), treatment should ideally be directed at improving the function of the heart as a pump. This will both increase cardiac output and decrease venous hypertension, thus improving both the low-output and congestive signs and symptoms of the disease. Unfortunately, this goal of improving the contractile state of the heart is often difficult to accomplish. Moreover, the chronic use of positive inotropic agents to improve contractility consistently increases mortality when compared to standard heart failure therapy.13-15 At the present time, standard pharmacologic management of heart failure is aimed at reducing ventricular preload and afterload and at diminishing, inhibiting, and/or antagonizing neurohormonal vasoconstrictor activation, rather than directly increasing cardiac contractility.
Prevention of Heart Failure
Typically, stage A and stage B patients have one or more of the following risk factors for the development of heart failure: advanced age, hypertension, diabetes mellitus, obesity, metabolic syndrome, coronary artery disease, prior myocardial infarction, and left ventricular hypertrophy. Other risk factors for heart failure include anemia, dyslipidemia, sleep disordered breathing, valvular heart disease, family history of cardiomyopathy, exposure to various cardiotoxins (eg, anthracyclines), skeletal myopathies, and other less common disorders (Table 28–2).
| Hypertension | Rheumatic fever |
| Diabetes | Mediastinal irradiation |
| Dyslipidemia | Sleep disordered breathing |
| Coronary artery disease | Collagen vascular disease |
| Valvular heart disease | Anemia |
| Obesity | Nutritional deficiencies |
| Metabolic syndrome | Exposure to cardiotoxic agents |
| Excessive alcohol consumption | Skeletal myopathies |
| Smoking | Thyroid disorders |
| Aging | Family history of cardiomyopathy |
Advancing age is one of the most prominent risk factors for heart failure. Data from the Framingham study indicate that heart failure is present in about 1% of adults in their 50s and in as many as 10% of those in their 80s.16 This association between advanced age and heart failure is most likely the result of decades of inadequate treatment of underlying risk factors, rather than to aging alone. Major risk factors for heart failure include hypertension, diabetes mellitus, coronary artery disease, prior myocardial infarction, and obesity.17 Obesity acts directly or indirectly in producing dyslipidemia, hypertension, insulin resistance, diabetes, and left ventricular hypertrophy, thus promoting heart failure.18 In this regard, the metabolic syndrome may comprise one of the most prevalent unifying risk factors for heart failure. Obesity may also contribute to the prevalence of obstructive sleep apnea, another independent risk factor for the development of hypertension and heart failure.19,20
As noted in Chap. 68, approximately 25% of the US population is hypertensive, and the lifetime risk of developing hypertension among Americans exceeds 75%.21 Elevated levels of systolic and/or diastolic blood pressure are major risk factors for the development of heart failure.22,23 Information from the Framingham Study suggests that hypertension precedes the onset of heart failure in 91% of cases.22 Thus, it is not surprising that long-term treatment of both systolic and diastolic hypertension has been shown to reduce the risk of heart failure.24,25 Numerous randomized controlled trials have consistently demonstrated that optimal blood pressure control decreases the risk of new-onset heart failure by approximately half.26
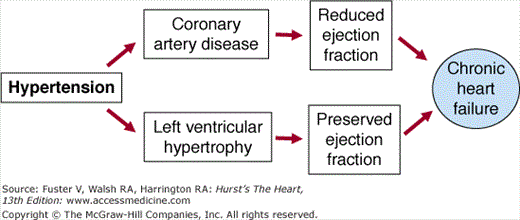
Hypertension may lead to structural heart disease and cardiac failure through at least two pathways, the development of left ventricular hypertrophy generally resulting in heart failure associated with a preserved left ventricular ejection fraction and myocardial infarction usually resulting in a regional wall motion abnormality and reduced left ventricular ejection fraction (Fig. 28–1). Left ventricular hypertrophy is a strong and independent risk factor for heart failure.27 However, in many instances, heart failure associated with a history of hypertension is mediated by atherosclerotic coronary artery disease rather than or in addition to left ventricular hypertrophy. In this circumstance, hypertension may be seen as a contributor to rather than a direct cause of the left ventricular dysfunction, which may be primarily attributed to myocardial ischemia/infarction. However, the role of hypertension leading to heart failure in postmyocardial infarction subjects should not be underestimated, as the benefits of treating hypertension in heart attack survivors are dramatic with an 81% reduction in the incidence of heart failure.24
To prevent heart failure and other cardiovascular events in hypertensive subjects, both systolic and diastolic blood pressure should be lowered using evidence-based, guideline-recommended therapies. Recommendations for the treatment of hypertension are reviewed in Chap. 70 and in the current version of the guideline from the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7).28 Goal blood pressure and the selection of an antihypertensive regimen depend, in part, on the absence or presence of comorbidity. Data support a lower ideal or target blood pressure goal in patients with associated major cardiovascular risk factors, particularly those with diabetes mellitus.29-31 While treatment to target blood pressure should remain the primary goal of antihypertensive treatment, the choice of specific agent(s) may be determined by the particular comorbidity present (eg, diabetes, coronary artery disease, or left ventricular dysfunction).
Various antihypertensive agents reduce the incidence of heart failure, including diuretics, angiotensin-converting enzyme (ACE) inhibitors, and β-blockers.28,32-34 Calcium channel blockers and α-blockers appear to be less effective in preventing the onset of heart failure. In fact, in the Antihypertensive and Lipid-lowering Treatment to Prevent Heart Attack Trial (ALLHAT), the α-receptor blocking agent doxazosin was associated with an increase in the incidence of new-onset heart failure.35 While the mechanism of increased heart failure risk with α-receptor blockade remains unknown, unopposed stimulation of β-adrenergic receptors is a likely possibility. In any event, α-blockade should be discouraged in the treatment of hypertension, unless in combination with other agents, particularly β-blockers. Finally, as noted in the European Society of Cardiology heart failure guideline,9 the applicability of ALLHAT to the European community is uncertain since the trial enrolled a substantial number of African Americans.
Like ACE inhibitors, angiotensin-receptor blockers (ARBs) significantly reduce the incidence of heart failure. In this regard, ARBs have been most well-studied in diabetic hypertensives.36,37 However, recent controversy has arisen regarding a so-called ARB paradox describing an apparent increase in myocardial infarction risk associated with ARB treatment.38,39 While this debate plays out, the totality of currently available data support the safety and efficacy of ARBs in the treatment of hypertension.
Many hypertensive subjects require treatment with two or more agents, to achieve target blood pressure control. With this in mind, the authors of the American College of Cardiology/American Heart Association heart failure guideline recommend the use of agents proven to be useful for the treatment of hypertension and heart failure, particularly diuretics, ACE inhibitors, and β-blockers, for the treatment of stage A and stage B hypertensive subjects.7 Likewise, the European Society of Cardiology endorses the use of ACE inhibitors, ARBs, diuretics, and β-blockers in the prevention of heart failure.9 Finally, the guideline from the Heart Failure Society of America emphasizes the use of an ACE inhibitor in stage A patients and the addition of a β-blocker in stage B subjects, particularly those with a prior myocardial infarction.8
Obesity and insulin resistance are powerful risk factors for the development of heart failure.40,41 Obesity alone has been shown to be an independent risk factor for incident heart failure.41 There are many ways by which obesity may contribute to the development of structural heart disease and heart failure. Many of these mechanisms seem to be mediated by the proliferative milieu associated with insulin resistance/hyperinsulinemia and diabetes. Thus, the metabolic syndrome may also play a major role in increasing the risk of heart failure.42 Approximately 40% of the US population older than 40 years of age meets the criteria for the diagnosis of the metabolic syndrome.43 Moreover, roughly 21% of US adults are obese as defined by a body mass index (BMI) of at least 30.44 This includes 20.8% of adult women and 21% of adult men for a total of 44.3 million obese adult Americans. Since 1991, the percentage of overweight adults (defined as a BMI ≥25) increased from 45% to 58%,44 so that more than one-half of the US adult population is now considered overweight. Unfortunately, other developed societies such as those in Europe are catching up to this high prevalence of obesity. Thus, the contribution of obesity, diabetes, and the metabolic syndrome to the incidence of heart failure is likely to increase, worldwide.
The presence of diabetes mellitus substantially increases the risk of heart failure in patients without preexisting structural heart disease.45 Interestingly, diabetes only modestly increases the risk of heart failure in men but substantially increases the chance of heart failure in women.22 While diabetes is a risk factor for coronary heart disease, many diabetic patients and especially female diabetics with or without hypertension exhibit angiographically normal epicardial coronary arteries in association with dilated cardiomyopathy. This observation has lead to the proposal of a diabetic cardiomyopathy. The exact nature of this form of heart muscle disease is unknown, but small vessel coronary disease and/or endothelial dysfunction may play a role. Alternatively, hyperinsulinemia, hyperglycemia, and other growth-promoting hormones may mediate pathological myocyte remodeling subsequently leading to cardiomyopathy in these patients.
The management of diabetes and the metabolic syndrome/obesity in the prevention of cardiovascular disease is discussed in Chaps. 91 and 92, respectively. In general, treatment of the metabolic syndrome is limited by the poor success associated with available weight loss therapies and programs. A decades-long downward trend in physical activity associated with an upward trend in caloric consumption and the lack of truly effective drug therapies challenge progress in effectively reversing the obesity epidemic.46 When successful, however, weight loss can effectively reduce the risk of diabetes in obese subjects. In the Diabetes Prevention Program (DPP), intensive lifestyle change, defined as a 7% reduction in body weight and at least 150 minutes of exercise weekly, was more effective than placebo or drug therapy with metformin in reducing the onset of diabetes mellitus.47 Lifestyle intervention decreased the incidence of diabetes by 58% compared to placebo, while metformin diminished the chance of diabetes by 31%.
In patients with overt diabetes, every effort should be made to control hyperglycemia, although such control has not yet been shown to reduce the subsequent risk of heart failure. Of note, thiazolidinediones (TZDs), commonly prescribed for the treatment of diabetes, have been associated with increased peripheral edema and new-onset heart failure in patients with underlying risk factors or known cardiovascular disease.48 The risk of developing edema with TZDs is dose related and is higher in diabetic patients on concomitant insulin therapy. Thus, these agents should be used with caution in at-risk patients. These patients should be monitored closely for fluid retention.
Coronary artery disease appears to account for 60% to 70% of the incidence of systolic heart failure. Annually in the United States, approximately 1.1 million individuals suffer a myocardial infarction and about 40% of them may be left with a reduced left ventricular ejection fraction.49 Data from the Studies of Left Ventricular Dysfunction (SOLVD) Registry, which enrolled subjects in the United States and Canada, provide the following breakdown on the etiology of heart failure: ischemic heart disease, 68.5%; idiopathic heart disease, 12.9%; hypertension, 7.2%; and other causes, 11.3%.50 This observation is supported by findings from randomized controlled trials of systolic heart failure that also demonstrate a preponderance of ischemic heart failure. However, these studies generally enrolled mostly middle-aged white men and may not have been representative of the broader heart failure population, including women and minorities. In this context, a recent trial of African American subjects (about one-half women) with systolic heart failure demonstrated a very different distribution of risk factors for heart failure, with only a minority of patients having ischemic heart disease and most exhibiting nonischemic heart failure in association with hypertension, diabetes, and obesity.51
The Acute Decompensated Heart Failure National Registry (ADHERE), which includes unselected patients admitted to the hospital with worsening heart failure, evaluated the prevalence of risk factors in more than 100,000 cases.52 One-half of the patients were women. The following predominant risk factors were noted: coronary artery disease, 57%; prior myocardial infarction, 35%; hypertension, 73%; dyslipidemia, 36%; chronic renal insufficiency, 30%; and diabetes mellitus, 44%. It is important to note that about 46% of ADHERE cases are comprised of patients with preserved left ventricular systolic function (ie, ejection fraction >40%). However even in the reduced left ventricular ejection fraction group, multiple comorbidities are common and ischemic heart disease is listed as the primary cause of the heart failure in only about 60% of cases.
In any event, myocardial infarction is a common cause of heart failure. Following a myocardial infarction, the development of left ventricular systolic dysfunction and dilation represent the most potent predictors of subsequent heart failure and all cause mortality.53-55 Once left ventricular injury has occurred, progressive left ventricular dysfunction and dilation ensues unless attenuated by medical and/or surgical therapy.
The pathophysiology and management of coronary artery disease is extensively reviewed in Part VIII. A consensus statement on preventing atherosclerotic heart disease provides additional guidelines for management.56 In this context, ACE inhibitors play a major role in the prevention of cardiovascular events and heart failure in subjects with established atherosclerotic vascular disease based on the results of randomized controlled trials.32 Revascularization strategies, either percutaneous or surgical, may also reduce the incidence of heart failure in such patients, although this is less well-studied in contemporary large-scale randomized controlled trials. However, coronary revascularization can relieve symptoms of myocardial ischemia, and coronary artery bypass surgery has been shown to lessen angina and reduce the risk of death in patients who have multivessel disease, reduced left ventricular ejection fractions, and stable angina.57 Whether or not surgical revascularization improves the natural history of ischemic heart failure is under evaluation in the National Institutes of Health sponsored Surgical Therapies for Ischemic Heart Failure (STICH) trial.
Other primary and secondary preventive strategies have also been shown to reduce the risk of incident heart failure. For example, lipid-lowering therapy results in a significant reduction in new-onset heart failure.58,59 In a large-scale trial, the administration of a lipid-lowering agent to patients with hypercholesterolemia and a prior myocardial infarction reduced all-cause mortality and the risk of developing heart failure.29 Thus, lipid-lowering therapy should play an appropriate role in any preventive strategy for heart failure. The role of other therapies such as continuous positive airway pressure breathing in obstructive sleep apnea to prevent new-onset heart failure remains to be fully elucidated. Other therapies resulting in reverse remodeling in patients with established (stage C) heart failure (eg, cardiac resynchronization therapy) are currently being evaluated in asymptomatic or minimally symptomatic patients, as preventive measures. In this regard, pathologic left ventricular remodeling has become a major target for heart failure intervention in stage B through stage D of the heart failure continuum.
As noted in practice guidelines, in the stage B patient preventive therapies should be directed at improving left ventricular structure and function. Typically, adverse or pathologic remodeling is defined in the context of left ventricular dilation and systolic dysfunction as an increase in ventricular volumes (both systolic and diastolic), a decrease in ejection fraction, a loss of the normal elliptical geometry of the ventricle, and the onset of functional mitral regurgitation. However, left ventricular hypertrophy should also be considered as a form of pathologic ventricular remodeling and as a target for preventive intervention. In this regard, the regression of left ventricular hypertrophy in hypertensive subjects appears to improve outcomes.60,61 Thus, therapies that promote either regression of left ventricular hypertrophy or reverse remodeling of the dilated failing heart may be viewed as essential in the prevention or attenuation of heart failure in stage B.
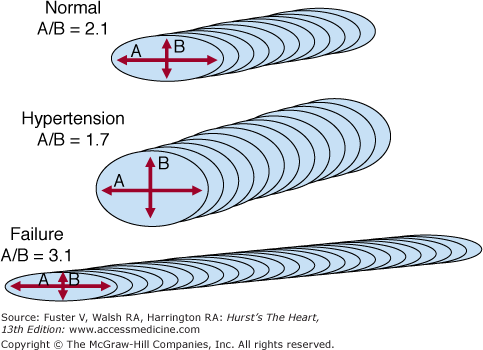
Pathologic ventricular remodeling occurs at three levels within the heart: the ventricular, cellular, and molecular levels. Ventricular remodeling is defined above. Cellular remodeling may take one of two predominant forms, involving either concentric or eccentric hypertrophy of the myocyte (Fig. 28–2).62 In the case of concentric myocyte remodeling, the heart cells have thickened but not increased in length so that the longitudinal to transverse ratio of the cell is diminished. With eccentric hypertrophy, the cell is elongated and the longitudinal to transverse ratio is increased. Other cellular changes accompanying pathological remodeling include changes in constituent cardiac proteins, an increase in the amount of cytoskeletal elements (however, these are disorganized), an increase in the number of nonmyocytes (many are activated [eg, fibroblasts] or dysfunctional [eg, endothelium]), and the extracellular matrix has increased in amount but is also disorganized. At the molecular level, there is a general reversion to the fetal pattern of gene expression in the dilated failing human heart.63,64 These changes appear to be related to increased wall stress and mediated by a variety of mechanisms, including increases in local and circulating neurohormones.65
Many drug therapies and some device-base treatments promote an improvement in cardiac structure and function. Most antihypertensive medications promote regression of left ventricular hypertrophy, although some appear more potent than others when indexed to the degree of blood pressure lowering.66 In subjects with asymptomatic left ventricular systolic dysfunction, the use of ACE inhibitors and β-blockers has been shown to be particularly effective in promoting reverse remodeling and attenuating clinical events.67,68 Angiotensin-receptor blockers and aldosterone antagonists may also be useful, in this regard. However, they have generally been studied in symptomatic patients with reduced ejections fractions including those who were recently postmyocardial infarction.69,70 Despite the lack of randomized controlled trials evaluating ARBs or aldosterone antagonists in truly asymptomatic left ventricular dysfunction, ARBs represent acceptable alternatives in ACE inhibitor–intolerant patients, and the addition of an aldosterone antagonist to an ACE inhibitor-/β-blocker-based regimen may optimize the reverse remodeling process. As mentioned above, mechanical interventions such as coronary revascularization following myocardial infarction or cardiac resynchronization therapy may also improve heart function and thus attenuate heart failure.
In summary, the best way to prevent heart failure is to aggressively treat major risk factors, including hypertension, diabetes, obesity, metabolic syndrome, and coronary artery disease, according to published guidelines. The treatment of other comorbid risk factors, such as dyslipidemia, should not be neglected as incremental reduction in heart failure risk may be seen. Once these risk factors progress to left ventricular hypertrophy or dysfunction, risk factor modification should continue and therapies that promote regression of left ventricular hypertrophy or reverse remodeling of the dilated failing heart should be employed, if not already prescribed.
Evaluation and Management of Symptomatic Heart Failure
Tables 28–3 and 28–4, taken from the European Society of Cardiology heart failure guideline,9 recommend a routine assessment to establish the diagnosis and likely cause of heart failure. Once the diagnosis of heart failure has been made, the first step in evaluating heart failure is to determine the severity and type of cardiac dysfunction, by measuring ejection fraction through two-dimensional echocardiography and/or radionuclide ventriculography. Measurement of ejection fraction is the gold standard for differentiating between the two forms of heart failure, systolic and diastolic, and is particularly important given that the approaches to therapy for each syndrome differ somewhat. The history and physical examination should include assessment of symptoms, functional capacity, and fluid retention. Common symptoms of heart failure are dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, cough (usually worse at night), abdominal complaints (nausea, vomiting, right upper quadrant pain, abdominal swelling), fatigue, nocturia, sleep disorders, and anorexia. Common physical findings include elevated jugular venous pressure, hepatojugular reflux, pulmonary crackles, sustained and displaced apical impulse, S3 gallop, hepatomegaly, ascites, and peripheral edema.
| Diagnosis of Heart Failure | ||||
|---|---|---|---|---|
| Assessments | Necessary | Supports | Opposes | Suggests Alternative or Additional Diagnosis |
| Appropriate symptoms | +++ | +++ (if absent) | ||
| Appropriate signs | +++ | + (if absent) | ||
| Cardiac dysfunction on imaging (usually echocardiography) | +++ | +++ (if absent) | ||
| Response of symptoms or signs to therapy | +++ | +++ (if absent) | ||
| ECG | +++ (if normal) | |||
| Chest x-ray | If pulmonary congestion or cardiomegaly | + (if normal) | Pulmonary disease | |
| Full blood count | Anemia/secondary polycythaemia | |||
| Biochemistry and urinalysis | Renal or hepatic disease/diabetes | |||
| Plasma concentration of natriuretic peptides in untreated patients (where available) | + (if elevated) | +++ (if normal) | Can be normal in treated patients | |
| Diagnosis of Heart Failure | |||
|---|---|---|---|
| Tests | Supports | Opposes | Suggests Alternative or Additional Diagnosis |
| Exercise test | + (if impaired) | +++ (if normal) | |
| Pulmonary function tests | Pulmonary disease | ||
| Thyroid function tests | Thyroid disease | ||
| Invasive investigation and angiography | Coronary artery disease, ischemia | ||
| Cardiac output | +++ (if depressed at rest) | +++ (if normal; especially during exercise) | |
| Left atrial pressure (pulmonary capillary wedge pressure) | +++ (if elevated at rest) | +++ (if normal; in absence of therapy) | |
Functional capacity is measured through history taking or preferably an exercise test.71,72 Analysis of expired air during exercise offers a precise measure of the patient’s physical limitations. However, this test is uncommonly performed outside of cardiac transplant centers. The NYHA has classified heart failure into four functional classes that may be determined by history taking:73class I, no limitations of physical activity, no symptoms with ordinary activities; class II, slight limitation, symptoms with ordinary activities; class III, marked limitation, symptoms with less than ordinary activities; and, class IV, severe limitation, symptoms of heart failure at rest. For example, patients who can walk several blocks without symptoms but have difficulty climbing two flights of stairs may have class II heart failure, while those who cannot walk several blocks easily or become winded while walking up a short flight of stairs might be considered class III. The NYHA functional classification should not be confused with the aforementioned stages of heart failure described in the American College of Cardiology/American Heart Association heart failure guideline. The NYHA classification describes functional limitation and is applicable to stage B through stage D patients, whereas the staging system describes disease progression somewhat independently of functional status.
Assessment of fluid retention through measurement of jugular venous pressure, auscultation of the lungs, and examination for peripheral edema is central to the physical examination of heart failure patients. The physical examination alone, however, is relatively insensitive for measuring extracellular fluid volume excess.74,75 A thorough appraisal of symptoms is therefore essential. Even in the absence of perceptible volume excess by physical examination, mild congestive symptoms can indicate volume excess. In addition, the chest x-ray, particularly in a patient with relatively new onset heart failure, is a relatively sensitive measure of volume overload. However, in the setting of chronic heart failure, the chest film may not reliably help estimate ventricular filling pressure.76
Given the limitations of physical signs and symptoms in evaluating heart failure clinical status, a number of noninvasive and invasive tools are under development for the assessment of heart failure. One such tool that has proven useful in determining the diagnosis and prognosis of heart failure is the measurement of plasma B-type natriuretic peptide (BNP) levels. Multiple studies, including the Breathing Not Properly Multinational Study,77 demonstrate the utility of BNP measurement in the diagnosis of heart failure. In this study, 1586 patients who came to the emergency department with acute dyspnea underwent bedside measurement of plasma BNP concentration. The clinical diagnosis of congestive heart failure was adjudicated by two independent cardiologists, who were blinded to the results of the BNP assay. The final diagnosis was dyspnea due to worsening heart failure in 744 patients (47%), dyspnea due to noncardiac causes in 72 patients with a history of left ventricular dysfunction (5%), and no finding of congestive heart failure in 770 patients (49%). B-type natriuretic peptide levels by themselves were more accurate than any historical or physical findings or laboratory values in identifying worsening heart failure as the cause of dyspnea. The diagnostic accuracy of BNP at a cutoff of 100 pg/mL was 83.4 percent. The negative predictive value of BNP was excellent. At levels less than 50 pg/mL, the negative predictive value of the assay was 96%.
In the B-Type Natriuretic Peptide for Acute Shortness of Breath Evaluation (BASEL) Study, from Mueller et al,78 patients presenting to the emergency department with acute dyspnea were randomly assigned to undergo either a single measurement of BNP or not. Based largely on the findings of the BNP Multinational Study, clinicians were advised that a plasma BNP concentration below 100 pg/mL made the diagnosis of congestive heart failure unlikely, while a level above 500 pg/mL made it highly likely. For BNP levels between 100 pg/mL and 500 pg/mL, the use of clinical judgment and additional testing were encouraged. In this single-blind trial of 452 patients, measurement of BNP in the emergency department resulted in a 10% decrease in the rate of hospital admission. Moreover, the median length of stay was reduced by 3 days and the mean total cost of treatment by about $1800. These reductions in rate of hospitalization, length of stay, and cost did not result in any negative effects on mortality or the rate of subsequent hospitalization.
Additionally, plasma BNP is useful in predicting prognosis in heart failure patients. However, serial measurement of plasma BNP as a guide to heart failure management has not yet been proven useful in the management of acute or chronic heart failure. The recommended role of BNP testing at the various stages of heart failure differs a bit among the major heart failure guidelines. The American College of Cardiology/American Heart Association heart failure guideline suggests that “measurement of BNP can be useful in the evaluation of patients presenting in the urgent care setting in whom the diagnosis of heart failure is uncertain.“7 Although the Heart Failure Society of America guideline discourages the use of BNP testing in “at risk“ patients, it more strongly recommends “that BNP or N-terminal-proBNP (NT-proBNP) be assessed in all patients (emphasis added) suspected of having heart failure when the diagnosis is not certain.“8 The heart failure guideline from the European Society of Cardiology emphasizes the use of BNP or NT-proBNP “as ‘rule out’ tests to exclude significant cardiac disease,“ particularly in primary care but also in settings such as the emergency department.9
Table 28–5 presents a general approach to evaluating and managing patients with current or prior symptoms of heart failure and chronic left ventricular systolic dysfunction. This schema is consistent with published clinical practice guidelines.7-10 In addition to pharmacologic and device-based approaches to treatment, these guidelines stress the importance of dietary counseling, patient education, lifestyle changes, and other nonpharmacologic strategies for the treatment of chronic heart failure. Many of these nonpharmacologic strategies recommended for the management of systolic heart failure also make good sense in the treatment of heart failure associated with a preserved left ventricular ejection fraction.
| Determine the etiology |
| Look for precipitating factors and correct them |
| Nonpharmacologic treatment |
| Sodium restriction (≤ 2 g/d) |
| Aerobic exercise |
| Weight loss in obese patients |
| Treat hypertension and other comorbidities vigorously |
| Pharmacologic treatment |
| Angiotensin-converting enzyme inhibitors (ACEIs) |
| Angiotensin receptor blockers (ARBs) |
| Aldosterone antagonists |
| β-Blockers |
| Vasodilators (long-acting nitrates and hydralazine) |
| Diuretics |
| Digoxin |
| Device therapies |
| Cardiac resynchronization therapy (CRT) |
| Implantable cardioverter defibrillators (ICDs) |
| Left ventricular assist devices (LVADs) (discussed in Chap. 30) |
When a patient presents with symptomatic heart failure, it is important to determine the underlying cause as some forms of heart failure may be reversible. It is also essential to understand those factors precipitating the transition from stage B to stage C heart failure, as well as subsequent episodes of decompensation. Therapy may then logically follow from an understanding of these factors and an appreciation for evidence-based, guidelines-recommended treatments.
All patients presenting with new heart failure should be rigorously evaluated in order to determine the etiology of the disease. Routinely performing such an evaluation is important because there are several surgically correctable forms of heart failure and some potentially reversible forms of inflammatory heart disease and cardiomyopathy associated with systemic diseases or cardiotoxins. For example, some patients with heart failure due to ischemic heart disease may realize an improvement in the left ventricular dysfunction and chronic heart failure following revascularization with either angioplasty or coronary artery bypass surgery.79-86 This may be the case in at least 20% of patients with ischemic heart failure, and as mentioned above, is being prospectively evaluated in the STICH trial. Thus, the presence or absence of obstructive coronary artery disease should be determined in heart failure patients and, in general, myocardial viability should be assessed in patients with ischemic cardiomyopathy. Viable myocardium and the presence of adequate target vessels for bypass predict a favorable outcome following revascularization for ischemic heart failure, so that serious consideration should be given to surgical intervention in such cases.
Perhaps the most classic examples of surgically correctable heart failure are those associated with valvular heart disease. For instance, patients with critical aortic stenosis may exhibit severe left ventricular dysfunction and advanced signs and symptoms of chronic heart failure which may be completely reversible upon replacement of the stenotic aortic valve. Severe mitral stenosis is also a cause of acutely reversible heart failure where percutaneous balloon valvuloplasty now represents a feasible and in many cases preferred approach to therapy.87 Severe mitral regurgitation is another valvular etiology of left heart failure. While early mitral valve repair or replacement may prevent the development of chronic heart failure, the presence of advanced left ventricular dilation and severe left ventricular dysfunction at the time of presentation often precludes surgical intervention. Newer, percutaneous approaches to mitral valve repair may represent a viable alternative to ongoing medical therapy in such patients.
The treatment of myocarditis remains controversial. Anecdotal experience suggests that some patients may benefit from therapy directed at the immune system. However, controlled trials of immunosuppressant therapy, immune globulin, and other forms of immunomodulatory therapy in myocarditis have been disappointing.88,89 Thus, current guidelines do not recommend the routine use of immune-based drug therapies for myocarditis. Despite these recommendations, many heart failure experts will recommend immunosuppressant drug therapy (usually consisting of corticosteroids) for biopsy-proven myocarditis associated with rapidly worsening heart failure signs and symptoms and/or left ventricular function. In this regard, the role of endomyocardial biopsy in the evaluation of heart failure remains controversial. None of the guidelines recommend the routine use of heart biopsies in the diagnosis of new or worsening heart failure. However, in highly suspected cases of myocarditis, endomyocardial biopsy can provide useful information establishing the diagnosis and distinguishing lymphocytic from eosinophilic and giant cell myocarditis. This is important because eosinophilic myocarditis is often exquisitely sensitive to treatment with corticosteroids, while giant cell myocarditis seems resistant to immunosuppressant treatment exhibiting a relapsing course despite such medications.
Cardiomyopathies related to alcohol or cocaine abuse may respond to discontinuation of the offending agent.90,91 Heart muscle disease associated with chemotherapeutic agents may respond to or be prevented by treatment with ACE inhibitors and/or β-blockers.92,93 Heart failure related to systemic illness such as hypothyroidism may respond to treatment of the underlying disease. While few specific therapies exist, the diagnosis of an infiltrative cardiomyopathy may influence subsequent evaluation for systemic disease and inform the natural history of the disease. A further discussion of these less common causes of cardiomyopathy is included in Chaps. 34 and 35.
One of the most important strategies for reducing morbidity and mortality in chronic heart failure is understanding and correcting the precipitating causes of clinical worsening and disease progression (Table 28–6). Dietary and/or pharmacologic noncompliance appear to be major causes of decompensation in patients with chronic heart failure. Thus, dietary counseling and patient education represent two major goals in the management of chronic heart failure. This may be accomplished through referral to the dietitian, distribution of patient education materials, office or hospital-based patient education programs, heart failure telemanagement, and home health cardiac specialists. Although an important first step, education delivered in the hospital alone is inadequate and this educational message must be reinforced on an outpatient basis.
| Dietary and/or pharmacologic noncompliance |
| Negative inotropes |
| Antiarrhythmics |
| First-generation calcium channel antagonists |
| Increased renal salt and water retention and/or worsening renal function |
| Nonsteroidal anti-inflammatory drugs (NSAIDs) |
| Further damage to the myocardium |
| Adriamycin |
| Alcohol and/or cocaine |
| Myocardial infarction |
| Myocarditis |
| Increased myocardial workload |
| Anemia |
| Hypoxia |
| Infection |
| Pulmonary embolism |
| Worsened valvular dysfunction |
| Arrhythmias |
The prescription of negative inotropes, such as most antiarrhythmics and the first-generation calcium channel antagonists, may also precipitate symptomatic or worsening heart failure and probably worsen outcome in chronic heart failure patients. Since renal perfusion in heart failure patients is largely dependent on vasodilating prostaglandins, the prescription or over-the-counter use of nonsteroidal anti-inflammatory drugs (NSAIDS) may result in worsening renal function and increased renal sodium and water retention.94 In addition, the onset of acute anuric renal failure may occur following the administration of NSAIDS to patients with advanced heart failure.95 Thus, some patients may make the transition from stage B to stage C heart failure as a consequence of inappropriate drug therapy.
Anything which further damages the myocardium, such as a new myocardial infarction, or which places an increased workload on the heart such as hypoxia, anemia, pulmonary embolism, or infection may result in worsening of heart failure. Finally, worsened valvular dysfunction and arrhythmias are occasionally implicated as precipitants of symptomatic heart failure.
The nonpharmacologic management of heart failure includes reduced sodium intake, reduced physical (particularly isometric) exertion, and weight loss in obese patients. In general, sodium intake should be limited to about 2 g/d. In advanced heart failure, further dietary sodium restriction may be necessary in order to attenuate expansion of extracellular fluid volume and the development of edema. Hyponatremia should not discourage compliance with a restricted sodium diet, since the hyponatremia is usually dilutional and associated with total body sodium and water excess. Sodium repletion therefore should only be considered in overt cases of severe excessive diuresis or dehydration. Salt substitutes may be used to improve the palatability of food. However, some of these agents substitute potassium chloride for sodium chloride, and should be used in moderation given the potential risk of hyperkalemia, especially in patients treated with ACE inhibitors, ARBs, and/or aldosterone antagonists. While dietary sodium restriction may attenuate the development of edema, it cannot prevent it since the kidneys are capable of reducing urinary sodium excretion to less than 10 mmol/d. Thus, diuretics (discussed below in Pharmacologic Treatment of Chronic Systolic Heart Failure) play a key role in the management of symptomatic heart failure.
While vigorous isometric exercise has been poorly evaluated in chronic heart failure, it is currently discouraged in symptomatic patients given the marked increase in myocardial work seen with weight training. Increased left ventricular wall stress during isometric exercise would be expected to have an unfavorable influence on ventricular remodeling. On the other hand, a role has emerged for monitored cardiac rehabilitation and aerobic exercise in patients with stable compensated chronic heart failure. In these patients, aerobic exercise training has been shown to consistently improve functional capacity while inconsistently improving left ventricular function, suggesting that the benefits of aerobic training in chronic heart failure are mostly peripheral.96-101 The effect of such training on heart failure outcomes is being evaluated in a large multinational National Institutes of Health randomized controlled trial.
While many patients with chronic heart failure have normal or low blood pressures, a large subset of patients remains substantially hypertensive. Since blood pressure is a component of ventricular afterload and it increases the workload of the heart, it should be treated vigorously in patients with chronic heart failure. Some patients with cardiomyopathy on the basis of hypertensive heart disease may recognize a marked improvement with treatment of the hypertension alone. This may be accomplished with standard heart failure treatment such as an ACE inhibitor or with various antihypertensive therapies which appear to be safe to use in patients with left ventricular systolic dysfunction (eg, vascular-selective calcium channel blockers). Other comorbidities, such as those mentioned earlier in this chapter, should also be treated and may modify disease progression as discussed above.
The current pharmacologic treatment of chronic systolic heart failure includes diuretics, digoxin, vasodilators, and neurohormonal inhibitors and antagonists.
In patients with heart failure, diuretics decrease end-diastolic volume and modestly increase stroke volume and cardiac output. Clinically, diuretics improve exercise capacity and diminish symptoms caused by pulmonary and peripheral edema. Figure 28–3 shows the tubular sites of action of commonly used diuretics and Table 28–7 reviews usual dosage recommendations for these agents.
| Agent | Site of Action | Total Daily Dose (mg) | Frequency |
|---|---|---|---|
| Acetazolamide | Proximal tubule | 250-375 | Every day or every other day |
| Amiloride | Distal tubule (potassium-sparing) | 5-20 | Once or twice daily |
| Bumetanide | Loop of Henle | 0.5-10 | Once to thrice daily |
| Chlorothiazide | Distal tubule (potassium-wasting) | 500-1500 | Once or twice daily |
| Ethacrynic acid | Loop of Henle | 50-200 | Once or twice daily |
| Furosemide | Loop of Henle | 20-500 | Once to thrice daily |
| Hydrochlorothiazide | Distal tubule (potassium-wasting) | 25-100 | Once or twice daily |
| Metolazone | Distal tubule (potassium-wasting) | 2.5-20 | Once or twice daily |
| Spironolactone | Distal tubule (potassium-sparing) | 25-200 | Once to thrice daily |
| Torsemide | Loop of Henle | 10-200 | Once or twice daily |
| Triamterene | Distal tubule (potassium-sparing) | 100-300 | Once or twice daily |
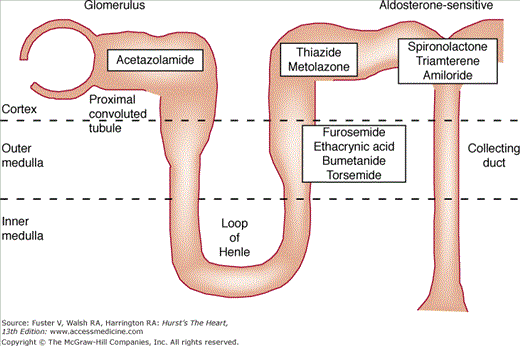
Diuretics that act primarily to decrease proximal tubular sodium reabsorption include osmotic diuretics such as mannitol, carbonic anhydrase inhibitors including acetazolamide, and probably the organomercurials. Except for organomercurials, the proximal tubular diuretics are not very effective when administered alone. Although 50% to 70% of the glomerular filtrate is reabsorbed isosmotically in the proximal tubule, the distal nephron particularly the ascending limb of Henle loop has the capacity to increase its rate of sodium reabsorption substantially.102 Therefore, an increase in glomerular filtration or decrease in proximal tubular reabsorption alone may not be associated with a significant diuresis, since the increased distal sodium and fluid delivery may be reabsorbed at more distal nephron sites. Thus, these agents are rarely used in clinical medicine and almost never used in the management of chronic heart failure.
Currently available loop diuretics include ethacrynic acid, furosemide, bumetanide, and torsemide. These are the most potent diuretic agents available for use in clinical medicine. Inhibition by these agents of active sodium chloride transport in the medullary ascending limb of Henle loop exceeds the rate-limited sodium chloride reabsorption in the more distal nephron, producing a maximal diuretic effect equivalent to 20% to 25% of the filtered sodium load. Accordingly, loop diuretics are most commonly used in the management of fluid retention in heart failure.
Distal tubular diuretics are broadly classified into two groups: the potassium-wasting and the potassium-sparing diuretics. Commonly used potassium-wasting distal tubular diuretics include the thiazide diuretics, chlorthalidone, and metolazone. These agents are chemically different but have similar diuretic effects. They inhibit only urinary diluting capacity and not concentrating capacity, since these distal tubular diuretics decrease sodium reabsorption in the cortical, but not medullary, segment of the ascending limb of Henle loop and the distal convoluted tubule. These distal tubular diuretics act proximal to the distal site of potassium secretion and thus are associated with an increase in urinary potassium excretion. This increase in potassium excretion appears to be due to an increase in distal sodium delivery which may modulate potassium reabsorption in the collecting duct.103 This is also the likely explanation for the potassium-losing effects of the loop diuretics.
Clinically-available potassium-sparing diuretics include triamterene, amiloride, and the specific aldosterone antagonists, spironolactone and eplerenone. The action of these latter potassium-conserving diuretics is dependent on the presence of the adrenal cortex and circulating aldosterone. In contrast, the ability of triamterene and amiloride to block potassium secretion is independent of adrenal function. Often, these diuretics are not potent enough when used alone, but may be used to avoid the potassium-losing effects of diuretics that act at more proximal nephron sites.
In contrast to the saluretic agents mentioned above, two currently available diuretics, demeclocycline and lithium, act at the level of the collecting duct and induce a water diuresis. These agents impair the ability of arginine vasopressin (AVP) to increase the water permeability of the renal collecting duct epithelium, thereby antagonizing the hydro-osmotic effect of AVP. Such agents are theoretically useful in managing the dilutional hyponatremia of heart failure; however, both of these agents produce significant adverse effects which preclude their use in heart failure management. Renal or V2 receptor AVP antagonists which directly antagonize the renal effects of AVP in the kidney offer a promising alternative to these agents and are currently undergoing human investigation.
With optimal dosing, all of the thiazide-like agents have generally comparable effects on solute and water excretion with the exception of metolazone which is more potent than the other agents. Thiazide diuretics differ from each other primarily in duration of action. In general, the loop diuretics are six to eight times more potent than the thiazides. This is not surprising, since several times more sodium chloride is reabsorbed in the loop of Henle than in the distal convoluted tubule. Because of their potency, the loop diuretics and metolazone are generally effective in patients with advanced renal insufficiency (ie, with glomerular filtration rates less than 25 mL/min). This is in contrast to the thiazide diuretics which become ineffective as the glomerular filtration rate falls below 25 to 30 mL/min.
During chronic administration, diuretic therapy results in favorable effects on cardiac preload and perhaps afterload with an associated improvement in LV performance. Wilson et al104 administered oral furosemide, with or without a thiazide, to 13 patients with hemodynamically decompensated heart failure. Right heart hemodynamics were determined at baseline and following 8 days of diuretic therapy. All patients exhibited a substantial decrease in left ventricular filling pressure which was generally associated with an improvement in cardiac performance measured by increases in stroke volume. Clinically, the favorable hemodynamic effects observed during chronic diuretic administration are associated with an improvement in functional capacity determined by treadmill exercise time. In a study from Bayliss et al,105 the administration of furosemide 40 mg plus amiloride 5 mg each daily for 1 month produced a nearly two-fold improvement in submaximal treadmill exercise time.
Chronic oral diuretic therapy has been shown to increase plasma renin activity, plasma angiotensin II concentration, plasma aldosterone, and plasma norepinephrine in the setting of acute and chronic heart failure.105,106 In addition to the diuretic-mediated increase in neurohormonal vasoconstrictor activation, chronic diuretic use also diminishes circulating concentrations of the vasodilating natriuretic peptides.106 This is important since natriuretic peptides are known to suppress the renin-angiotensin-aldosterone system and to improve renal hemodynamics. Moreover, during chronic diuretic therapy, further activation of vasoconstrictor/anti-natriuretic forces and diminution of vasodilating/ natriuretic plasma hormone concentrations may explain, in part, the development of progressive diuretic resistance which is common in chronic heart failure.
Volume depletion and potassium depletion are the most common complications of diuretic therapy. Thiazide diuretics, metolazone, and loop diuretics commonly cause these complications. Volume depletion may result in hypotension and/or hypoperfusion of vital organs such as the brain, heart, or liver. Diminished renal perfusion also may occur, as evidenced by a rise in blood urea nitrogen and serum creatinine concentrations, despite ongoing hypervolemia. Generally, one-third of patients undergoing treatment for acutely decompensated heart failure experience a worsening of renal function.107 Severe renal hypoperfusion may lead to the evolution of acute intrinsic renal failure (ie, acute tubular necrosis), in addition to the development of severe prerenal azotemia. Hypokalemia may predispose heart failure patients to life-threatening ventricular arrhythmias, particularly during treatment with cardiac glycosides or other positive inotropes.
Hyponatremia may result from the impaired water excretion associated with chronic heart failure. In addition, the saluretic effect of commonly prescribed diuretics may produce or exacerbate hyponatremia, especially in patients with advanced heart failure permitted ad libitum fluid intake. Symptomatic hyponatremia is better treated by water restriction than by cessation of diuretic therapy, unless overdiuresis or dehydration has occurred. Metabolic acidosis is a complication of the use of carbonic anhydrase inhibition, since these agents block hydrogen ion secretion. The use of thiazide and loop diuretics may also cause metabolic alkalosis. This is usually due to the excretion of sodium, chloride, and potassium without bicarbonate, which leads to a rise in serum bicarbonate concentration. Other adverse effects of diuretics include carbohydrate intolerance, hyperuricemia, hypersensitivity reactions (interstitial nephritis, skin rashes, hematologic disorders), and acute pancreatitis. Deafness, which is generally reversible upon cessation of diuretic administration, may be associated with the use of ethacrynic acid or furosemide. Unfortunately, some cases of diuretic-induced deafness are not reversible. Finally, one of the most perplexing and challenging adverse effects of diuretics is the development of diuretic resistance. While no generally agreed upon definition exists, diuretic resistance is generally manifest as worsening renal function during diuresis or a diminishing/inadequate response to diuretic administration. This clinical situation has also been described as a part of the so-called cardiorenal syndrome. Importantly, diuretic resistance or the cardiorenal syndrome has been associated with worse outcomes (increased morbidity and mortality) in both chronic and acutely decompensated heart failure.108-110
The mechanisms leading to diuretic resistance are many. Some degree of renal insufficiency is usually present in patients with chronic heart failure. This insufficiency is related to renal hypoperfusion, which affects the amount of diuretic delivered to the tubular lumen of the kidney. With a decreased level of diuretic being delivered to its site of action, a decrease in drug potency is present. Additionally, though loop diuretics are used in various stages of renal insufficiency, thiazide diuretics have a much narrower window of efficacy with worsening renal function as mentioned above.111 Also, inadequate dosing of diuretics can affect their efficacy. For example, the duration of action of furosemide is approximately 6 to 7 hours. Thus, a prolonged period for “compensatory“ sodium retention occurs during the balance of the 24-hour period, when this diuretic is administered on a once-daily regimen. Dietary control of sodium intake is essential in mitigating this effect.112
The chronic use of loop diuretics can also lead to a braking phenomenon, which stimulates anatomical and functional alterations of the distal tubule. The resultant hypertrophy of these segments leads to an increased reabsorptive ability of the tubules and overall decreased natriuresis.113
Patients with advanced heart failure often develop fluid deposition in a number of varying body sites, including the gastrointestinal tract. Gut edema can often prevent the proper absorption of orally administered diuretic agents, thereby delaying the time to achieve a peak dose effect. As noted, a lack of dietary control, namely with salt intake, can result in a net positive sodium balance, despite increasing diuretic doses.114 The use of NSAIDs can also be linked to diuretic resistance. Also as mentioned, inhibition of cyclooxygenase by these agents results in decreased renal prostaglandin synthesis. Prostaglandins play a crucial role in not only offsetting the systemic increase of vasoconstrictors induced by heart failure but also by promoting natriuresis.115 Use of these agents can thereby decrease the effectiveness of administered diuretics and also potentially contribute to a nephrotoxic state.
Finally, diuretic therapy via several of the aforementioned mechanisms may initiate an iatrogenic cardiorenal syndrome leading to worse heart failure outcomes (Fig. 28–4).
Figure 28–4.
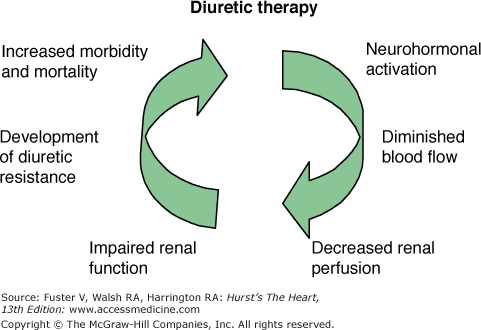
The iatrogenic cardiorenal of heart failure. Diuretics further activate neurohormonal vasoconstrictor systems resulting in an impairment in renal function and diuretic resistance by the mechanism shown. Diuretic resistance has been associated with poor outcomes in acute and chronic heart failure.
Before a patient can be deemed as diuretic resistant, a number of factors should be taken into account. Chief among them should be drug compliance, as this would be the first area of concern if any resistant state should be questioned. If compliance with medications is not an issue, then sodium and fluid restriction should also be closely monitored, as this area is also notoriously difficult to successfully manage in the outpatient heart failure setting. Concomitant medications, such as NSAIDs, should have their use curtailed or stopped if possible. Additionally, medications that serve to antagonize the hormonal cascade induced by the renin-angiotensin-aldosterone and adrenergic nervous systems, such as ACE inhibitors and β-blockers, should be optimized to their fullest extent.
After these factors are thoroughly evaluated and adjusted, the alteration of diuretic administration can be attempted. A common method to address an inadequate diuretic response in a patient who is on an oral loop diuretic is to increase the amount of diuretic dosage. As mentioned, decreased renal perfusion can diminish the secretion of loop diuretic into the renal tubule; thus, the use of an increased dose of a loop diuretic is a relatively easy way to increase the amount of drug delivered to the nephron.116 Another variation is to increase the frequency of diuretic dosing, as this approach minimizes the compensatory postdiuretic salt retention that commonly occurs after loop diuretic administration, given the relatively short half-lives of these agents.
Loop diuretics target only specific areas of the nephron, and are thus unable to alter sodium reabsorption elsewhere in the renal circuit. Indeed, their long-term use can facilitate increased distal tubular sodium and water reabsorption. Combination therapy with other classes of diuretics can be an additional method of maximizing diuresis, through the blockade of multiple tubular sites of sodium reabsorption. Though proximally-acting diuretics, such as acetazolamide, and potassium-sparing diuretics, such as spironolactone, can be utilized for this purpose, their chronic use brings into play a number of safety issues (eg, metabolic acidosis and hyperkalemia, respectively). Spironolactone has established itself as a valued medication for chronic heart failure use, but this is based more on its antagonism of aldosterone as opposed to its diuretic potency. Thiazide diuretics, on the other hand, have emerged as viable additions to loop diuretic therapy. This approach to sequentially block sodium reabsorption at numerous sites of the nephron has proven to be effective, although signs of overdiuresis and electrolyte depletion should be carefully monitored.117,118 Metolazone, a thiazide-like diuretic, had received an initial push as the agent of choice to increase diuresis in resistant patients taking a loop diuretic because of its greater potency than other thiazides. However, hypokalemia and rapid volume depletion are real risks of this approach; alternatively, hydrochlorothiazide therapy is quite effective and carries with it less risk of producing these untoward effects.119-120
Diuretic therapy should be initiated with a loop or thiazide-type diuretic, based on the severity of the heart failure. For severe heart failure with overt volume overload (ie, pulmonary and/or peripheral edema), the more potent loop diuretics (furosemide, ethacrynic acid, bumetanide, and torsemide) should be used. If a loop diuretic given twice daily in doses equivalent to furosemide 100 to 200 mg/d is inadequate for diuresis, a thiazide diuretic or metolazone may be added. This often results in a synergistic effect on solute and water excretion, as noted above. Since this combination also results in substantial renal potassium wasting, an increased need for potassium supplementation should be anticipated. In order to conserve potassium and/or to further enhance the diuresis, a potassium-sparing diuretic may be added. However, triamterene should not be combined with furosemide since triamterene blocks the tubular secretion of furosemide thus inhibiting furosemide effect.121 The goal of diuretic therapy in heart failure is resolution of the signs and symptoms caused by pulmonary and/or peripheral edema. Thus, once this goal has been achieved, the diuretic dose should be adjusted to maintain a euvolemic state. With specific tailored guidelines, patients may be instructed to alter their diuretic regimen on an as needed basis to alleviate signs and symptoms of extracellular fluid volume excess.
ACE inhibitors are among the mainstays of heart failure therapy. In patients with chronic heart failure, ACE inhibition improves hemodynamics and functional status (ie, lessens symptoms and improves exercise tolerance), reduces hospitalizations for heart failure, and prolongs life.
ACE inhibition exerts a number of beneficial hemodynamic effects in patients with chronic heart failure. These effects include decreases in systemic vascular resistance, pulmonary capillary wedge pressure, right atrial pressure, and end-diastolic and end-systolic dimensions and improvements in cardiac performance as evidenced by increased cardiac output and stoke volume and by improved fractional shortening determined by echocardiography. The acute and chronic hemodynamic effects of ACE inhibition in heart failure were first described in the early 1980s. LeJemtel et al122 examined the acute hemodynamic response to a single oral 25 mg dose of the ACE inhibitor, captopril, in patients with established heart failure. At baseline, these patients exhibited severely decompensated heart failure with an average systemic vascular resistance of nearly 2000 dynes/s/cm5, pulmonary capillary wedge pressure of almost 30 mm Hg, and cardiac index of just 2 L/min/m2. Following the administration of the ACE inhibitor, both systemic vascular resistance and pulmonary capillary wedge pressure fell significantly. This was associated with a marked improvement in the cardiac index.
DiCarlo et al123 investigated the acute and chronic hemodynamic effects of ACE inhibition in patients with chronic heart failure. Patients were given the oral ACE inhibitor enalapril at a dose of 10 mg twice daily. Hemodynamics were assessed prior to treatment, following the initial 10 mg dose, and after 30 days of chronic therapy. Both the acute and chronic administration of the ACE inhibitor improved hemodynamics, with significant decreases in systemic vascular resistance and pulmonary capillary wedge pressure and increases in cardiac index.
Chronic ACE inhibition also improves left ventricular dimensions and function in patients with chronic heart failure. This was demonstrated in 1982 by Awan et al124 who examined the effects of ACE inhibition on measures of left ventricular size and performance as determined by echocardiography in nine patients with chronic heart failure. Left ventricular end-diastolic and end-systolic dimensions and shortening fraction were determined at baseline and after 1 week, 8 weeks, and 6 months of chronic ACE inhibition. Through 8 weeks of therapy, there was a progressive decrease in left ventricular dimensions and a statistically significant improvement in shortening fraction, evident at 1 week and maintained through 6 months of therapy.
The effects of ACE inhibition on symptoms and on exercise capacity in chronic heart failure include improvements in symptoms of heart failure determined by patients’subjective impressions (eg, quality of life, patient global assessments), NYHA functional class ranking, and increased exercise tolerance times. Thus, ACE inhibitors improve both subjective and objective measures of functional capacity in chronic heart failure.
In 1984, Sharpe et al125 examined the effect of chronic ACE inhibition on heart failure symptoms as reported by the patient. Thirty patients were randomized to receive either the ACE inhibitor enalapril (10 mg twice daily) or a matching placebo for 3 months. All 16 patients randomized to the ACE inhibitor reported feeling “much better“ at follow-up. In contrast, only 3 of 14 placebo-treated patients reported feeling “much better.“ In the placebo group, five patients report feeling “little better,“ two patients reported “no change,“ three patients reported feeling “little worse,“ and one patient reported feeling “much worse.“ Four patients in the placebo group died prior to the end-of-study assessment. A number of other trials in chronic heart failure have also demonstrated improvement in symptoms and in NYHA functional class ranking in patients randomized to ACE inhibitors.126-133 For example, the Captopril Multicenter Research Group examined the effect of chronic ACE inhibition on NYHA functional classification in a 12-week, double-blind, placebo-controlled evaluation of 74 patients with established heart failure.132 While 61% of the 46 ACE inhibitor-treated patients demonstrated improvement in NYHA functional class ranking, only 24% of placebo-treated patients exhibited functional class improvement. This ability of chronic ACE inhibition to improve NYHA functional class ranking was seen at the end of 2 weeks of study, was progressive to 8 weeks of therapy, and then maintained throughout the remainder of the 12-week period of study.
The beneficial effects of chronic ACE inhibition on exercise capacity in patients with chronic heart failure are also well established.132,134,135 In the aforementioned Captopril Multicenter Research Group trial, treadmill exercise time was significantly and progressively improved throughout the 12 weeks of study in patients randomized to the ACE inhibitor.132 There was a 24% improvement in exercise duration in the ACE inhibitor group, and virtually no change in the placebo group. Chalmers et al135 demonstrated similar findings using bicycle exercise testing and another ACE inhibitor, lisinopril. In this randomized, double-blind, placebo-controlled investigation, a significant 200-second improvement in bicycle exercise time was seen by 4 weeks of study and maintained through the end of study (week 12) in patients randomized to the ACE inhibitor.
ACE inhibition has been shown to: (1) prolong survival in advanced, NYHA class IV, heart failure; (2) prolong survival in mild to moderate, NYHA class II and class III, heart failure; and, (3) prolong survival in patient with asymptomatic LV dysfunction following a myocardial infarction and when heart failure complicates an acute myocardial infarction. A discussion of ACE inhibition in postmyocardial infarction patients is beyond the scope of this chapter and is discussed in Chaps. 61 and 64.
The first survival trial of ACE inhibition in chronic heart failure was the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS), published in 1987.136 In this double-blinded clinical trial, 253 patients with advanced heart failure were randomized to receive either placebo or the ACE inhibitor enalapril. The average enalapril dose was 18.5 mg/d or about 10 mg given twice daily. ACE inhibition produced a highly significant reduction in mortality throughout the 1-year period of follow-up. Following the CONSENSUS trial, the Studies of Left Ventricular Dysfunction (SOLVD) investigators examined the survival benefit of ACE inhibition in patients with mild to moderate chronic heart failure.137 More than 2500 patients were randomized in this double-blinded, placebo-controlled trial of enalapril. During the 4-year period of follow-up, there was a highly significant 16% reduction in mortality observed in the ACE inhibitor group. These proven effects of ACE inhibition to prolong survival in NYHA class II through class IV patients support their use as first-line agents in the pharmacologic management of chronic heart failure.
All three heart failure guidelines strongly endorse the use of ACE inhibitors as first-line agents in the management of all NYHA classes of chronic systolic heart failure. In the absence of fluid retention, these agents should be initiated first in the management of systolic heart failure, and in the case of those with fluid retention, an ACE inhibitor should be started concomitantly with a diuretic. ACE inhibitors should be initiated at a low dose and slowly titrated to doses proven to be effective in randomized controlled trials. Virtually all ACE inhibitors evaluated in heart failure have proven effective; Table 28–8 lists the evidence-based ACE inhibitors used in heart failure, including their starting and target doses. Patients should be monitored carefully for the major side effects of ACE inhibition including hypotension, prerenal azotemia, hyperkalemia, and cough. Cough in a heart failure patient taking an ACE inhibitor should not immediately be presumed related to the medication, as elevated left ventricular filling pressure may commonly cause coughing. In this instance, the patient may need an increase in the diuretic dose or an increase in rather than a discontinuation of the ACE inhibitor. Finally, angioedema is a rare adverse effect of ACE-inhibitor therapy.
| Agent | Total Daily Dose (mg) | Route of Administration | Frequency |
|---|---|---|---|
| Captopril | 75-150 | Oral | Thrice daily |
| Enalapril | 10-40 | Oral | Twice daily |
| Enalaprilat | 2.5-20 | Intravenous | Every 6 h |
| Fosinopril | 10-40 | Oral | Once daily |
| Lisinopril | 10-40 | Oral | Once daily |
| Quinapril | 10-40 | Oral | Once or twice daily |
| Ramipril | 2.5-20 | Oral | Once or twice daily |
| Trandolapril | 1-4 | Oral | Once daily |
Angiotensin receptor blockers (ARBs) have been evaluated for use as either alternatives to ACE inhibitors or as additive therapy to an ACE-inhibitor- and β-blocker-based heart failure drug regimen. The concept of enhanced blockade of the renin-angiotensin system with the addition of an angiotensin II receptor (AT1) blocking agent is conceptionally appealing and has been evaluated in multiple randomized controlled trials. Theoretically, blockade of the AT1 receptor should result in more complete antagonism of the renin-angiotensin system and act independently of non-ACE pathways for angiotensin II production. Alternatively, blocking the effects of angiotensin II at the receptor level rather than inhibiting its production via ACE inhibition could act to preserve the postulated benefits of AT2-receptor agonism. On the other hand, ACE inhibition affects not only the renin-angiotensin pathway but also the degradation of the vasodilator, bradykinin. Specifically, ACE inhibition results in decreased bradykinin degradation and thus increased circulating levels of bradykinin. Hence, the mechanisms of action of ACE inhibitors versus ARBs are complex and can be used to argue for the theoretical superiority of either agent over the other.
The effects of ARBs on symptoms, exercise capacity, and systemic hemodynamics in heart failure appear to be similar to those observed with ACE inhibitors.138-140 Angiotensin II receptor blockers are extremely well tolerated and do not cause cough. Some, early studies suggested that angiotensin II receptor blockers maintain or improve glomerular filtration, in contrast to the decrease in glomerular filtration commonly seen with ACE inhibitors.140-142 This proposed effect of angiotensin II receptor antagonists may be attributable to their lack of effect on the vasodilator bradykinin.142 However, as noted below, a differing effect between ACE inhibitors and ARBs on renal function has been difficult to demonstrate in clinical trials.
The Evaluation of Losartan in the Elderly (ELITE-I) trial hypothesized that the ARB, losartan, would be equally efficacious and associated with the development of less prerenal azotemia, in comparison with the ACE inhibitor, captopril.141 In fact, the incidence of worsening renal function defined by an increase in serum creatinine of at least 0.3 mg/dL was the same in both groups. Although ELITE-I failed to meet its primary renal function end point, it suggested the potential superiority of the ARB over the ACE inhibitor on the combined secondary end point of hospitalizations or mortality and on mortality alone. However, the ELITE-I trial was not prospectively designed or powered to assess morbidity or mortality, and at best, it may be viewed as hypothesis-generating.
Like ACE inhibitors, ARBs improve survival in heart failure patients when compared to placebo. The comparative efficacy of ACE inhibitors and ARBs on survival has been a topic of great interest over the past decade. The Losartan Heart Failure Survival Study (ELITE II) was designed to compare the efficacy of an ARB and an ACE inhibitor (again, losartan and captopril, respectively) in elderly subjects with symptomatic heart failure due to left ventricular systolic dysfunction (LV ejection fraction <40%).143 The study, designed as a follow-up to ELITE-I, evaluated all-cause mortality as its primary end point. There was no difference between these agents in all-cause mortality, thus failing to confirm any superiority of ARBs in the treatment of heart failure. Moreover, this trial was not statistically powered to determine the equivalency of the two agents, leaving the role of ARBs in the treatment of heart failure somewhat uncertain. More recently, a trial in patients with left ventricular dysfunction following acute myocardial infarction demonstrated the noninferiority of the ARB, valsartan, when compared to the ACE inhibitor, captopril.69 However, there was no incremental reduction in morbidity or mortality when the two agents were used in combination.
The utilization of ACE inhibitors and ARBs together in patients with chronic mild or moderate heart failure has been evaluated in two large-scale randomized controlled trials. The Valsartan Heart Failure Trial (Val-HeFT) enrolled 5010 patients to determine the effects of valsartan (target dose 160 mg twice a day) versus placebo added to an ACE inhibitor in patients with NYHA class II or class III heart failure.144 This study demonstrated a slight but statistically significant reduction in a clinical composite end point (all-cause mortality plus heart failure hospitalization) favoring the addition of the ARB. In addition, valsartan improved left ventricular ejection fraction and decreased left ventricular dimensions as measured by echocardiography. A post hoc subgroup analysis of 1610 patients on β-blocker and ACE inhibitor therapy at baseline, however, suggested an increase in mortality with the addition of valsartan (valsartan, 16.4% vs placebo, 12.5%; P = .018) raising concerns about the combination of ARB, ACE inhibitor, and β-blocker.
The Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) trial addressed this issue further by prospectively analyzing the effect of candesartan (target dose of 32 mg/d) versus placebo in 4576 patients with reduced left ventricular ejection fractions and mild or moderate heart failure. 145 (A separate cohort of patients with preserved left ventricular systolic function was also evaluated in the CHARM study program.) This study reported a statistically significant reduction in the combined end point of cardiovascular morbidity and mortality when candesartan was combined with an ACE inhibitor or an ACE inhibitor plus a β-blocker. The explanation for the disparity between Val-HeFT and CHARM in the “triple therapy“ group (ACE inhibitor-, ARB-, and β-blocker-treated patients) is unclear but could be related to differences between the two ARBs, different dosing strategies, or chance.146 In CHARM, the assessment of triple therapy was a prespecified end point, making this the more valid assessment of triple neurohormonal therapy with an ACE inhibitor, ARB, and β-blocker.
Thus, results from Val-HeFT and CHARM support the concept of a clinical benefit to more complete antagonism of the renin-angiotensin system. However, the magnitude of benefit is relatively small, about a 10% to 15% reduction in the combined end point of all-cause mortality and heart failure hospitalization. In both studies, the reduction in mortality alone with the addition of an ARB was less than 10% and was not statistically different from placebo. Finally, both trials identified a highly significant survival advantage of ARB administration in patients intolerant to an ACE inhibitor.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


