Depositional Diseases of the Lungs
INTRODUCTION
Deposits of endogenous body constituents or exogenous materials in amounts sufficient to deform structure and impair function can occur virtually anywhere in the body. Deposits of endogenous materials in the lungs or airways cause a variety of diseases (Table 64-1). These may have different clinical manifestations, depending on localization (i.e., pulmonary parenchyma or conducting airways). This chapter deals with a few of these manifestations: amyloidosis; diffuse pulmonary calcification; alveolar microlithiasis; diffuse alveolar hemorrhage (DAH) syndromes; and idiopathic pulmonary hemosiderosis. Others are discussed elsewhere in this text.
AMYLOIDOSIS
Characteristics of amyloid and the various forms of pulmonary amyloid are considered below.
 NATURE OF AMYLOID
NATURE OF AMYLOID
Amyloidosis refers to the extracellular deposition of amyloid, a fibrillar, proteinaceous, insoluble material that has characteristic light, ultrastructural, and histochemical features (Fig. 64-1). Electron microscopic examination of amyloid reveals a dominant (95%) fibrillar component with distinctive periodicity, associated with a lesser (5%) pentagonal doughnut-shaped glycoprotein component, physically and chemically identical in all forms of amyloid, which is derived from a soluble plasma protein, soluble amyloid P protein (SAP). Amyloid also includes various glycosaminoglycans and certain apolipoproteins (E and J). Radiographic diffraction studies of amyloid show the fibrils to be arrayed in a β-pleated sheet configuration. This accounts for the ordered binding of the histochemical stain Congo red such that Congo red–stained amyloid appears apple-green under polarized light.1,2 When amyloid is deposited in tissues it may produce atrophy of parenchymal cells (e.g., glomeruli), interference with mechanical function (e.g., heart and lungs), or impaired vasoconstriction of blood vessels, leading to hemorrhage (e.g., lungs and gastrointestinal tract). Other mechanisms of tissue injury are also hypothesized including direct tissue toxicity.3
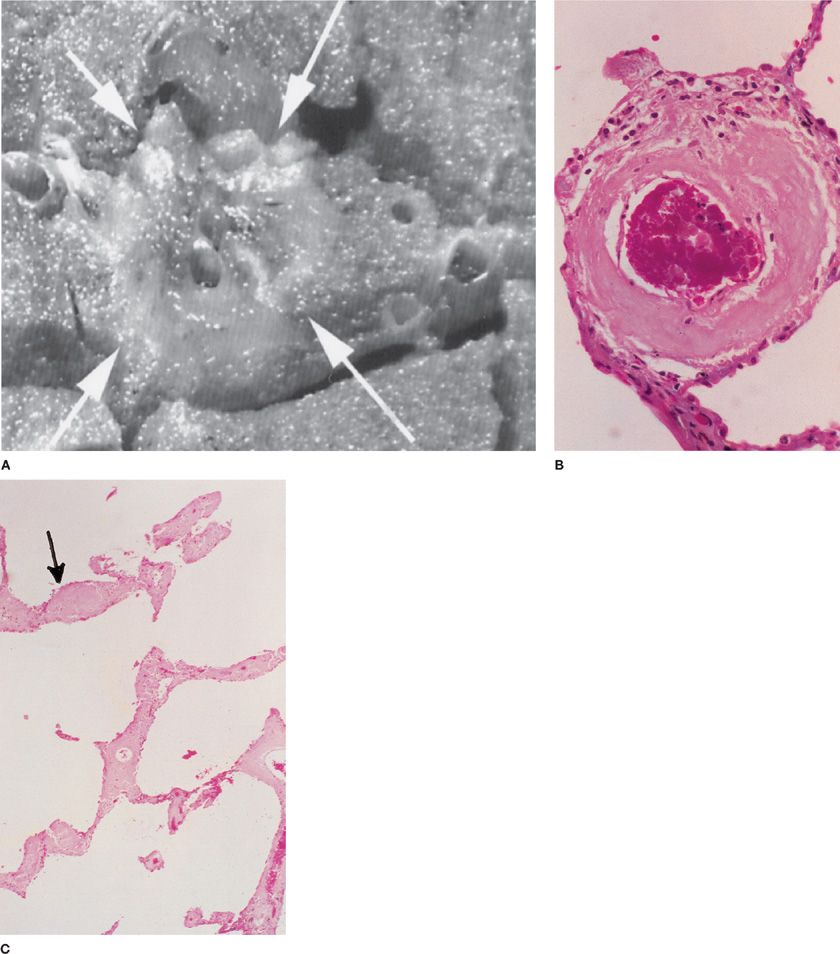
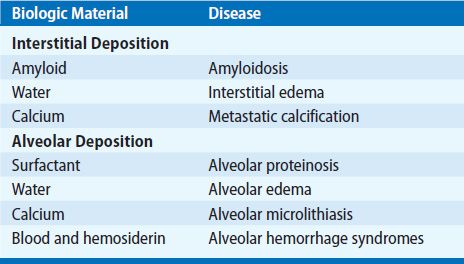
Figure 64-1 Amyloid deposition. A. Amyloidoma. Cut surface of lung with white arrows indicating a dense, wax-like lesion that is characteristic of nodular amyloid. Incidental finding at autopsy. (Used with permission of Leslie A. Litzky, MD, Department of Pathology and Laboratory Medicine, Hospital of the University of Pennsylvania, Philadelphia.) B. The typical amorphous appearance of amyloid is seen deposited within the wall of a pulmonary venule. Green birefringence on polarized light examination after staining with Congo red will confirm the amyloid nature of the deposit (H&E ×700). C. Amorphous amyloid in the alveolar interstitial space. Arrow indicates a thickened alveolar septum (H&E ×420).
While the main fibrillar component of amyloid can be derived from any one of 27 precursor proteins,2 only a few are common in systemic disease, including immunoglobulin light-chain, serum amyloid–associated (SAA) protein (a family of acute phase reactants), transthyretin (TTR, a prealbumin molecule that binds and transports thyroxine and retinol), and β2-microglobulin.1,4–6 The established amyloid fibril nomenclature is based on the chemical nature of the fibril protein, where an A, for amyloid, is followed by a suffix that is an abbreviated form of the parent or precursor protein. These forms of amyloidosis are therefore known as AL, AA, ATTR, and Aβ2M, respectively. An entity related to amyloidosis is light-chain deposition disease (LCDD) in which tissue deposits are also derived from immunoglobulin light chains and are similar to amyloid by light microscopy, but show granular deposition by electron microscopy and do not stain with Congo red.7 It seems likely that the biochemical properties of the light chain determine the nature of the deposit produced.
AL amyloidosis usually occurs in association with a neoplastic clonal proliferation of B cells or plasma cells which produce a monoclonal immunoglobulin or immunoglobulin fragment (monoclonal gammopathy). The neoplastic clone may clinically manifest as multiple myeloma or lymphoma (generally lymphoplasmacytic lymphoma) or may be subclinical (formerly known as primary amyloidosis), causing bone-marrow plasmacytosis. Most often the protein source is a λ-light chain, either intact or the amino terminal fragment. Amyloid-associated (AA) amyloidosis (previously referred to as secondary amyloidosis) is associated with a chronic increase in serum acute-phase reactants. It was formerly seen predominantly in patients with chronic infections (e.g., tuberculosis, leprosy, and chronic osteomyelitis) but is now seen more commonly with noninfectious chronic inflammatory diseases (e.g., rheumatoid arthritis, familial Mediterranean fever, Crohn disease, and heroin abuse with “skin popping”).1,6 TTR is deposited in familial amyloid polyneuropathies and senile systemic amyloidosis (ATTR amyloidosis).5 β2-microglobulin deposition (Aβ2M amyloidosis) is seen in patients with chronic renal failure on dialysis.
 PULMONARY INVOLVEMENT IN AMYLOIDOSIS
PULMONARY INVOLVEMENT IN AMYLOIDOSIS
It is important to distinguish secondary involvement of the respiratory tract in patients with systemic disease from localized pulmonary involvement with the latter being much less common than the former.4,8,9 Tracheobronchial amyloid deposition and nodular parenchymal amyloid deposition (amyloidoma) (Fig. 64-1A) most often occur as isolated phenomena, whereas diffuse interstitial deposition is more often seen in systemic amyloidosis. In addition to pulmonary involvement per se, amyloidosis may also cause symptoms in any portion of the respiratory tract or there may be secondary effects from deposition in other organs. For example, deposits in the tongue may be extensive enough to cause obstructive sleep apnea. Persistent pleural effusions may be due to both pleural and cardiac disease.10 Diaphragmatic deposition may lead to respiratory failure. Pulmonary hypertension is a rare complication.11 The vast majority of cases of pulmonary amyloidosis can be categorized as tracheobronchial amyloidosis, nodular parenchymal amyloidosis, and diffuse septal amyloidosis.
Nodular Parenchymal Amyloidosis
Solitary amyloid nodules (amyloidomas) are commonly incidental radiographic findings in asymptomatic individuals (Fig. 64-1).4,8 When multiple, such nodules may be associated with cough, dyspnea, or hemoptysis. These nodules have no distinctive features, although occasionally, they may show radiographic evidence of calcification or cavitation. Usually the diagnosis of an amyloid nodule is made after surgical resection. Occasionally, the diagnosis has been made by transbronchial biopsy or percutaneous fine-needle aspiration. However, surgical excision of one or more nodules may be prudent, since, on rare occasion, amyloid deposition occurs within a pulmonary neoplasm (e.g., a primary neoplasm such as atypical carcinoid or a metastatic neoplasm such as medullary carcinoma from the thyroid). It is possible that advances in biochemical analysis of the amyloid (see below) may influence this decision, but this is not yet commonly discussed in the literature.
Nodular parenchymal amyloidosis most often represents AL. Histologically, the amyloid deposit is often associated with an intense inflammatory reaction consisting of plasma cells, macrophages, and multinucleated giant cells. Interestingly, when the accompanying plasma cells have been analyzed for clonality, they are more often polyclonal than monoclonal. In such cases, the inflammatory cells may therefore be a local reaction to the presence of amyloid, rather than the source of the amyloid precursor light chains. In a few instances, nodular amyloidosis has been associated with a histologically apparent low-grade pulmonary lymphoma. Rare cases of AA amyloidosis have also been reported. Clinical follow-up of nodular parenchymal amyloidosis unassociated with systemic or, frankly, neoplastic disease is generally benign.8
Tracheobronchial Amyloidosis
Amyloid deposition in the tracheobronchial tree can produce either plaques or tumoral masses.4,9 The more common presentation as plaques is diffuse and multifocal, and represents submucosal deposition of amyloid. Diffuse or proximal involvement of the airways is apt to be symptomatic, producing cough, stridor, or hemoptysis. Less commonly, deposition of amyloid in the tracheobronchial tree produces a solitary mass, which mimics an endobronchial neoplasm with signs of bronchial obstruction or hemorrhage. Tracheobronchial amyloid deposition, like parenchymal nodule amyloid, is most often of light-chain derivation and a localized phenomenon. Again, like nodular amyloidosis, this form is rarely associated with systemic disease. Both types of airways lesions can be readily identified by bronchoscopic examination. However, as is the case with amyloid deposition at all sites, with biopsy there is a risk of hemorrhage. Although localized tumoral masses may be treated by excision or observation, more diffuse involvement may be treated by laser ablation, stents, or radiation. Involvement of proximal airway leads to significant mortality while involvement of distal or mid airway has generally good prognosis.
Diffuse Interstitial Amyloidosis
Widespread, diffuse interstitial amyloidosis of the pulmonary parenchyma may produce either a reticulonodular or miliary pattern on the chest radiograph.4,8,12 The deposition of amyloid may involve the alveolar septal interstitium, the walls of small blood vessels, or both (Fig. 64-1B,C). Such pulmonary involvement occurs most often in patients with systemic amyloidosis, derived from either immunoglobulin light-chain or AA protein. Pulmonary interstitial amyloid deposition in secondary involvement is rarely sufficiently severe to produce clinical manifestations but, uncommonly, it may produce progressive dyspnea, hemoptysis, or restrictive pulmonary function tests. It is thought that the morbidity and mortality of diffuse septal amyloidosis in patients with systemic disease is related to concurrent cardiac amyloidosis with which tissue burden closely correlates, possibly explaining why septal amyloidosis by itself in this setting is not of more significance.4 On the other hand, primary diffuse interstitial pulmonary amyloidosis has been considered to have a relatively poor prognosis.4,12
 DIAGNOSIS AND TREATMENT OF AMYLOIDOSIS
DIAGNOSIS AND TREATMENT OF AMYLOIDOSIS
Diagnosis of amyloidosis requires tissue examination and Congo-red staining and/or electron microscopy. The biochemical nature of the amyloid fibril in patients with systemic amyloidosis cannot be predicted from clinical manifestations alone.13–18 Even in patients with a known plasma cell dyscrasia, if amyloid is detected, it should not be assumed that this represents AL amyloid since a significant number of patients with familial or senile type amyloid also have plasma cell dyscrasias. Historically, a variety of immunohistochemical and immunofluorescence tests were used, combined with genetic testing; but these commonly required frozen tissue, which were technically challenging and did not always provide an answer. More recently, improved immunohistochemistry and biochemical techniques have become available and are applicable in formalin fixed paraffin embedded tissue.13–17,19–21 Published data show these approaches are highly robust and generalizable to multiple clinical scenarios although they have not yet been reported in patients with isolated pulmonary disease.
While it is beyond the scope of this chapter to discuss therapy in depth, it is important to note that therapy for systemic disease is entirely dependent on the specific peptide responsible. In cases of systemic AL amyloidosis, there has been considerable progress in treatment with introduction of myeloma type therapy with high-dose prednisone and mephalan therapy combined with use of hematopoietic stem-cell transplantation. Newer agents also appear promising such as thalidomide and related compounds and especially bortezomib which targets plasma cells.3 In contrast, treatment for AA amyloidosis focuses on the underlying inflammatory disease but may also include small molecule inhibitors.6 Systemic senile amyloidosis due to deposition of transthyretin (ATTR) has also been approached with small molecule inhibitors.5 In some cases of hereditary disease, either small molecule inhibitors or organ (liver, heart) transplantation has been explored.5 The significance of these therapies for lung-limited disease is unclear since localized disease is typically treated with ablative therapy such as stents, radiation, laser treatment, resection, and other localized methods. The more recent systemic therapies have not yet been reported in patients with lung-limited disease.
DIFFUSE PULMONARY CALCIFICATION
Calcification of the pulmonary parenchyma can occur by a variety of mechanisms.22 Dystrophic calcification refers to the deposition of calcium salts, most often crystalline hydroxyapatite, in dead tissue such as within the healing granulomas of tuberculosis or sarcoidosis. It can also be seen in various other conditions such as in pulmonary hypertension, other post infectious conditions or pneumoconiosis. This type of calcification is usually localized; its distinctive radiographic features are sometimes diagnostically helpful. It is rarely of any clinical significance beyond that of the underlying condition.
Metastatic calcification refers to the deposition of calcium salts, usually amorphous, in normal tissues (Fig. 64-2). This latter type of calcification occurs in association with some derangement of calcium metabolism, such as primary hyperparathyroidism, secondary hyperparathyroidism of chronic renal failure, hypervitaminosis D, the milk-alkali syndrome, sarcoidosis, or increased bone turnover due to multiple myeloma or metastatic carcinoma.
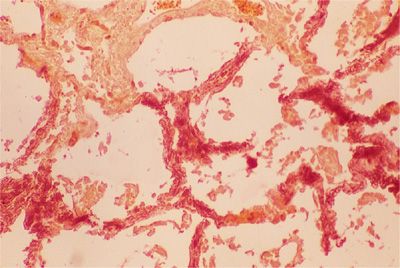
Figure 64-2 Metastatic calcification of alveolar septa in a renal dialysis patient. Photomicrograph shows calcium forming a dark red precipitate within the alveolar septa (Alizarin red ×280).
Although metastatic calcification can occur in almost any tissue of the body, it occurs most often in the lungs, kidneys, and the stomach (tissues with more alkaline pH), and the walls of blood vessels. Metastatic calcification in the lungs usually affects the interstitium of the alveolar septa and the walls of bronchioles and pulmonary vessels, sometimes localizing on elastic fibers.
Clinical manifestations of diffuse pulmonary calcification are unusual, occurring most often in patients who are in chronic renal failure, particularly in those on chronic hemodialysis. Radiographically, metastatic calcification usually takes the form of a diffuse interstitial infiltrate, sometimes with fine nodularity. Less often, confluent patchy consolidation mimicking pneumonia may be seen. Although the calcific nature of the infiltrate is often apparent on routine chest radiograph, computed tomography (CT) scan is more sensitive both in detecting the interstitial deposits and in revealing their calcific nature. Moreover, CT scan may also demonstrate calcification of chest wall blood vessels, circumstantially implicating calcification as the cause of pulmonary parenchymal abnormalities. Recognition of the calcific nature of the infiltrate is furthered by scanning with 99mtechnetium.
Only rarely do the patients manifest dyspnea or arterial hypoxemia, and pulmonary function tests tend to not show signs of restrictive pulmonary disease. Unexplained dyspnea in a patient with chronic renal failure or hypercalcemia in the presence of a normal chest radiograph should lead to consideration of high-resolution computed tomography (HRCT) or technetium scanning. Rarely, respiratory failure may develop although death from metastatic calcification is typically due to cardiac disease.
The mechanism responsible for diffuse pulmonary calcification is unknown. Although high levels of parathyroid hormone or a marked increase in the calcium-phosphate solubility product occur in some patients, diffuse calcification can occur in the absence of either. Ultrastructural observations of minimal, presumably early, lesions show selective deposition of calcium on elastic fibers, suggesting that they may serve as the initial nidus. In contrast to their apparent role in alveolar microlithiasis, extracellular matrix vesicles do not appear to be involved.
ALVEOLAR MICROLITHIASIS
Pulmonary alveolar microlithiasis (PAM) is a rare autosomal recessive disorder characterized by intra-alveolar accumulation of spherical calcified concretions (called calciferites, calcospherites, or microliths), in the absence of any known calcium metabolism disorder.23 This disorder usually presents with an abnormal chest radiograph from an asymptomatic patient (Fig. 64-3). Presentation can occur at any age, although symptoms usually occur in third or fourth decade of life. The chest radiograph and/or HRCT are diagnostic, showing a sand-like micronodulation throughout the lung fields. This is caused by the presence of innumerable minute calcified spherules filling the alveolar spaces. Although not usually required, bronchoalveolar lavage or biopsy can confirm the diagnosis. Biopsy shows calcified spherules filling alveolar spaces (Fig. 64-3).
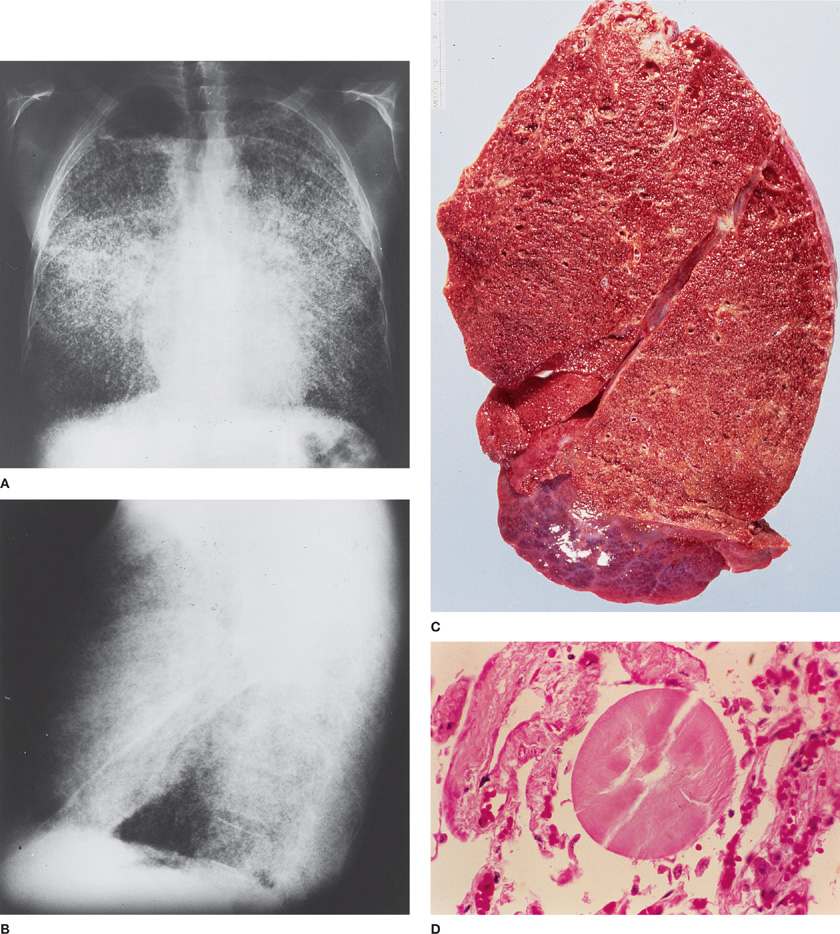
Figure 64-3 Alveolar microlithiasis in a 46-year-old man admitted for nonpulmonary problems. History included slight dyspnea on exertion and previous episodes of “pneumonia” in 1947, 1950, and 1952. Clinical examination revealed severe restrictive lung disease, pulmonary hypertension, and cor pulmonale. Diagnosis confirmed by lung biopsy. A and B. Posterior–anterior and lateral chest radiographs demonstrate innumerable, tiny calcified nodules throughout both lung fields. Thin, lucent lines on each side represent normal pleura visualized between the calcified pulmonary parenchyma and the chest wall. Emphysematous blebs in the apices displace the calcifications. C. Cut surface of explanted lung from a patient undergoing lung transplantation for primary alveolar microlithiasis. Note the fine nodularity which correlated with the chest radiographs. (Used with permission of Leslie A. Litzky, MD, Department of Pathology and Laboratory Medicine, Hospital of the University of Pennsylvania, Philadelphia.) D. Photomicrograph demonstrating a typical calcospherite in an alveolar space (H&E ×1120).
Although usually asymptomatic at the time of presentation, alveolar microlithiasis typically progresses to end-stage lung disease, but the rate of progression is highly variable. When it does, the findings are those of restrictive pulmonary disease or exercise-induced pulmonary hypertension. No therapy has proven effective but lung transplantation has been performed successfully in a few patients.
The disease is due to a mutation in a type IIb sodium-phosphate cotransporter (SLC34A2) gene. As surfactant phospholipids are metabolized, phosphate is liberated and the transporter is needed to eliminate the excess phosphate from the alveolar space. In the absence of this transporter, increased phosphate levels lead to microlith formation. While this is most dramatic in the lungs, other organs can also be affected. The accumulation of microliths ultimately leads to loss of vital capacity by mass effect as well as by inducing underlying parenchymal fibrosis.
ALVEOLAR HEMORRHAGE SYNDROMES
Pulmonary hemorrhage most commonly arises from endobronchial diseases (tumors, bronchiectasis, bronchitis).24 However, there is a subset of patients in whom bleeding originates at the level of the alveoli and who are referred to as having DAH.25 Symptoms range from cough, fever, and dyspnea alone to respiratory failure. While hemoptysis is common, it is not universal, even when DAH is severe. In these cases, the diagnosis can be suspected due to a falling hemoglobin level. Evaluation of serial bronchoalveolar lavage aliquots in such patients may show a progressive increase in bloody return, as opposed to endobronchial disease, in which bleeding tends to clear. Bronchoalveolar lavage is also useful to exclude infection in patients with DAH.
The damage to alveolar septa may either be due to immunologic mechanisms (immune complex, antineutrophil cytoplasmic antibody [ANCA], antiglomerular basement antibodies, antiphospholipid antibodies) or to nonimmunologic causes. This distinction is largely, although not perfectly, captured in the presence or absence of the pathologic finding of capillaritis (Fig. 64-4 and Table 64-2).26 Capillaritis is characterized by infiltration of alveolar walls by inflammatory cells, usually neutrophils, but sometimes eosinophils or monocytes, with fibrinoid necrosis of the alveolar and vessel wall. However, due to the absence of supporting structures, alveolar necrosis leads to wall breakdown so rapidly that this latter feature may be hard to appreciate. In order to distinguish this process from simple margination of neutrophils, there should be evidence for neutrophils undergoing apoptosis (pyknosis and nuclear fragments). Distinction from infection requires determination that there is minimal accumulation of inflammatory cells within alveoli. The pathologic diagnosis of pulmonary hemorrhage itself requires that there is either hemosiderin-laden macrophages or evidence of hemophagocytosis, since the blood that is commonly seen in lung biopsies may be due to surgery alone. If this evidence is absent, clinical criteria for DAH should be used.
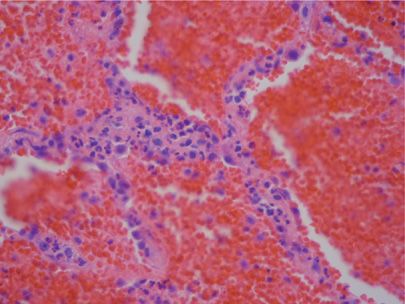
Figure 64-4 Capillaritis as a cause of pulmonary hemorrhage. Note the neutrophils in the alveolar septum (in center of image) and the blood and fibrin in the alveolar spaces (H&E ×100).



