Stable angina pectoris
Unstable angina pectoris
Variant angina (“Prinzmetal’s angina”)
Acute myocardial infarction
Non-Q wave or non-ST-elevation (usually nontransmural infarcts)
Q-wave or ST-segment elevation (usually transmural myocardial infarcts)
Angina pectoris is the clinical term used to describe chest discomfort resulting from a relative oxygen deficiency in heart muscle. Heberden [1] named this entity in 1786 when he identified a “disorder of the breast marked with strong and peculiar symptoms and considerable for the kind of danger belonging to it” associated with a “strangling and anxiety,” which he suggested should be called “angina pectoris.” Herrick [2] enlarged on this description in 1912. Angina is usually described by the patient as a left precordial tightness or ache provoked by exercise or emotion and relieved by rest. Many patients deny they have pain, but when questioned closely, they identify chest tightness, chest pressure, or aching associated with effort, emotion, and/or exposure to cold. Angina occurs when the oxygen demand exceeds the supply [3–7]. Most individuals with angina have underlying atherosclerotic coronary artery disease (CAD) (Figs. 15.1 and 15.2).
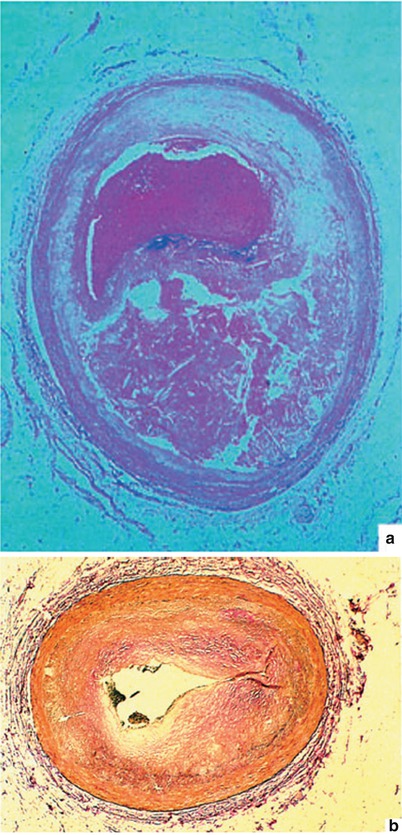
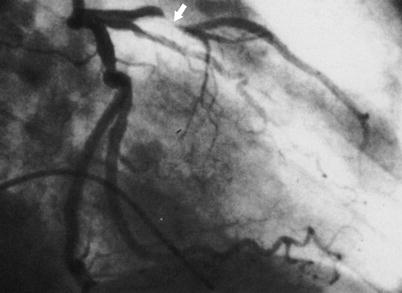
Fig. 15.1
(a) A typical atherosclerotic plaque that has ruptured, leading to the development of coronary artery thrombosis. In such a case, the patient may or may not have had angina at effort before the plaque rupture and thrombosis. He or she may have abruptly developed severe chest pain and a myocardial infarction with plaque rupture and coronary artery thrombosis. (b) Neointimal proliferation occurring with restenosis after coronary artery angioplasty, leading to coronary artery luminal diameter narrowing and the need for an additional revascularization procedure. Patients who develop coronary heart disease after cardiac transplantation also have this same type of alteration (i.e., neointimal proliferation) in the coronary arteries of their new heart. Native atheromas have substantial fibroproliferative alterations as well
Fig. 15.2
Coronary arteriogram of the left coronary artery in the right anterior oblique projection, showing typical narrowing and occlusion (arrow) of the coronary artery by atherosclerosis, as typical in patients with unstable angina and myocardial infarction. In many patients, the left anterior descending coronary artery stenosis is less severe than that shown here
However, angina may also develop in some patients with ventricular hypertrophy, left ventricular (LV) outflow obstruction, severe aortic valvular regurgitation or stenosis, cardiomyopathy, or dilated ventricle(s) in whom severe coronary artery stenoses are not present. When CAD is not present, the explanation for angina is that even normal coronary arteries may not adequately supply hypertrophied, dilated, or failing heart muscle with oxygen. On occasion, limited coronary vasodilator reserve may explain angina, especially in some patients with severe ventricular hypertrophy, in patients with LV hypertrophy associated with LV outflow obstruction, including valvular aortic stenosis and hypertrophic obstructive cardiomyopathy, and in patients with severe LV hypertrophy associated with systemic arterial hypertension [8, 9]. Coronary artery vasoconstriction occurring with exercise or stress may also be a contributing factor [10, 11]. Most humans without significant coronary heart disease or ventricular hypertrophy do not develop angina with effort or stress, probably because the heart is protected from a serious imbalance between oxygen supply and demand by other factors that limit physical activity, such as dyspnea and fatigue.
The predisposing pathologic alteration in coronary arteries responsible for angina is atherosclerosis, atherothrombosclerosis, and neointimal fibrous proliferation (Fig. 15.3; see also Figs. 15.1 and 15.2). The term atherothrombosclerosis reflects the relative importance of each of its components, including atherosclerosis, thrombosis, and fibrosclerosis, in the development of the process that has long been called atherosclerosis but that, in fact, often includes evidence of thrombosis in the progressive atherosclerotic plaque. Embolic atherosclerotic debris and platelet aggregates may contribute to distal coronary artery occlusion and limitation of coronary flow reserve (Fig. 15.3). Severe narrowing of the lumen of the coronary artery results in a decreased ability to deliver oxygen, especially when oxygen demand in the heart is increased, as with increases in heart rate, contractile state, myocardial wall tension, or a combination of these factors [3–7]. Therefore, stable and relatively predictable angina may develop during exercise, exposure to cold, or emotional stress or after eating a large meal. Angina may also occur because of extracardiac influences. In particular, severe anemia or carbon monoxide exposure reduces the amount of oxygen delivered to the heart and may result in angina under conditions that would otherwise be well tolerated. Increases in systemic arterial pressure and consequent dilatation of the heart may result in angina. Increases in the heart rate or contractile state, as with hyperthyroidism, pheochromocytoma, and exogenous administration or release of catecholamines, may also lead to angina. Exposure to cold decreases oxygen delivery by causing coronary artery vasoconstriction and increased systemic blood pressure, ventricular wall tension, and oxygen demand.
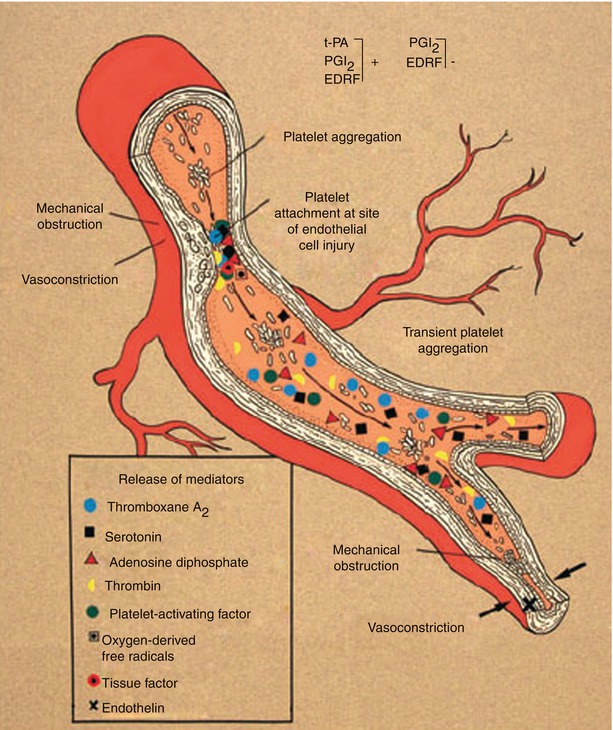
Fig. 15.3
Schematic diagram suggesting probable mechanisms responsible for the conversion from chronic coronary heart disease to acute coronary artery disease syndromes. In this scheme, an endothelial injury, generally plaque ulceration or fissuring at the site of an atherosclerotic plaque, is associated with platelet adhesion and aggregation and with the release and activation of selected mediators, including thromboxane A2, serotonin, adenosine diphosphate, platelet activating factor, thrombin, oxygen-derived free radicals, and endothelin. Local accumulation of thromboxane A2, serotonin, platelet activating factor, thrombin, adenosine diphosphate, and tissue factor promotes platelet aggregation. Thromboxane A2, serotonin, thrombin, and platelet activating factor are vasoconstrictors at sites of endothelial injury. Therefore, the conversion from chronic stable to acute unstable coronary heart disease syndromes is usually associated with endothelial injury, platelet aggregation, accumulation of platelet and other cell-derived mediators, further platelet aggregation, and vasoconstriction, with consequent dynamic narrowing of the coronary artery lumen. In addition to atherosclerotic plaque fissuring or ulceration, other reasons for endothelial injury include flow shear stress, hypertension, immune complex deposition and complement activation, infection, and mechanical injury to the endothelium as it occurs with coronary artery angioplasty, stents, and after heart transplantation. Injured endothelial cells have reduced amounts of the normally present vasoprotective substances that, when present, prevent thrombosis, vasoconstriction, and inflammation; these substances include nitric oxide, tissue-type plasminogen activator (t-PA), and prostacyclin (PGI 2 ). EDRF denotes endothelin-derived relaxing factor, which is nitric oxide
Physical Examination/Bedside Findings
On physical examination, the findings in patients with stable angina pectoris are highly variable. Often, there are no localizing or suggestive physical findings. Alternatively, associated risk factors, such as systemic arterial hypertension, hyperlipidemia, valvular heart disease, heart failure, or peripheral atherosclerosis, may result in a specific physical finding, such as elevated blood pressure, a prominent fourth heart sound (S4), an accentuated aortic closure sound, a paradoxically split second heart sound (S2) [systemic arterial hypertension, during an episode of angina or with left bundle branch block (LBBB)], a reduced peripheral pulse (i.e., carotid, femoral, or lower-extremity pulse with or without a bruit over the artery), a murmur of aortic stenosis or mitral insufficiency (the valvular heart diseases most commonly associated with CAD), or a third heart sound (S3) with heart failure or rapid filling of the ventricle, such as occurs with moderately severe and severe aortic or mitral insufficiency. The patient with coronary heart disease and stable angina may or may not have an enlarged heart; frequent or complex ventricular premature beats; S4, a murmur of mitral insufficiency; S3; moist rales; and peripheral vascular disease.
The electrocardiogram (ECG) may be normal, but it often shows ST-T wave changes, usually ST-segment depression and T-wave flattening or inversion during angina. On occasion, ST-T-wave depression or inverted T waves persist for weeks or longer, reflecting chronic ischemia. The ECG may also show ventricular ectopic beats or evidence of a previous Q-wave myocardial infarction (MI). An echocardiogram may be normal at rest or may show regional or global wall motion abnormalities, including a reduced LV ejection fraction or increased LV dimensions (“ischemic cardiomyopathy”), caused by myocardial ischemia or infarction or by mitral insufficiency. Chest radiographs may show a normal-sized or enlarged heart with or without heart failure. On occasion, the patient has visible coronary artery calcification on the chest radiograph (see section “Coronary artery calcification scoring”).
Hemodynamic monitoring typically shows increases in the mean pulmonary capillary wedge pressure during angina pectoris. With the onset of myocardial ischemia, the initial hemodynamic change in the left ventricle is a decrease in myocardial compliance and an increase in stiffness. This results in a sharp increase in the mean pulmonary capillary wedge pressure during angina, with a return to baseline as the angina resolves. The change in compliance is followed by ST-T wave changes on the ECG, a decline in regional systolic wall thickening, and, finally, the development of chest pain. This entire sequence occurs within a few seconds.
Coronary Artery Calcification Scoring
Coronary artery calcification scoring by means of fluoroscopy, electron beam (EB) computed tomography (CT), multidetector (MD) CT, or coronary computed tomography angiography (CCTA) can improve risk stratification, especially in patients at intermediate risk [12–15]. In symptomatic patients, the presence of coronary calcium entails at least a fourfold increased risk of death or infarction compared with an absence of calcification. Indeed, a high calcium score has been shown to be predictive of the presence of advanced coronary plaque and stenosis. In general, greater degrees of calcification in coronary arteries are consistent with greater amounts of atherosclerotic plaque and more advanced coronary luminal diameter narrowing. An advantage of coronary artery calcium assessment is that it can be done irrespective of the patient’s ability to exercise and of the presence or absence of resting electrocardiographic abnormalities. A valuable finding in the symptomatic patient is a zero or very low coronary calcium score (i.e., <10). The negative predictive value of such a calcium score for significant stenosis of a major coronary artery is greater than 90 % with EBCT or MDCT and nearly 100 % with CCTA. Therefore, one of these methods might be an appropriate first test in individuals with atypical cardiac symptoms in whom the likelihood for ischemic disease is considered to be small by the responsible physician. In the United States, more than 8 million patients with chest pain present to emergency departments each year [13, 16]. Most of these patients are admitted to the hospital for 24–48 h to rule out ACS, but only 2–15 % of them are actually diagnosed with this condition [15, 16]. As a first-line imaging test for low- to intermediate-risk patients with acute chest pain, CCTA not only detects ACS when present but also appears to allow safe, expedited discharge of patients who have negative test results [13–15] (Fig. 15.4). These patients may generally be reassured and further testing directed at noncardiac etiologies of chest pain. On the other hand, if the calcium score is consistent with moderate or severe atherosclerotic plaque development, additional cardiac evaluation, including stress testing (ideally with perfusion or functional evaluation), may be indicated. It is important to keep individual variation in mind, however, as coronary heart disease may be the cause of chest pain even in the absence of coronary artery calcification.
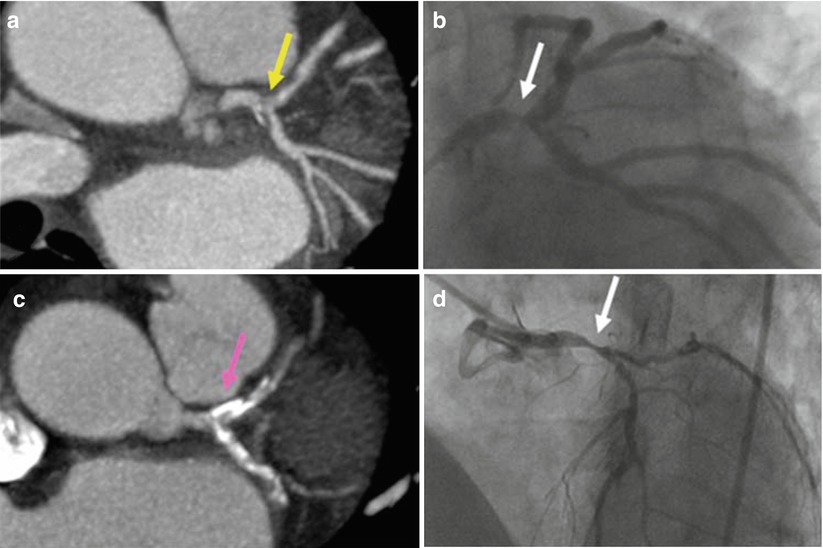
Fig. 15.4
Computed coronary tomography angiography (CCTA). An area of non-calcified plaque seen on CCTA (a) corresponds with a high grade left main coronary artery stenosis (yellow arrow) identified with conventional x-ray angiography (b). In a different patient, several areas of calcification (such as the one indicated by the white arrow) in the left main, proximal left anterior descending, and proximal left circumflex arteries are identified on CCTA (c) but preclude the diagnosis of a high-grade stenosis (the pink arrow indicates the area where the narrowing exists but is not visible on CCTA), which was subsequently seen (white arrow) on conventional angiography (d) (From Cyrus et al. [17]. Reprinted with permission from Springer)
Asymptomatic individuals differ from symptomatic patients in that the risk of subsequent morbid events is relatively small in the absence of symptoms. However, several recent studies have indicated that coronary artery calcium scoring may be useful for evaluating asymptomatic patients at intermediate risk for CAD [18–22]. As Fig. 15.5 shows, prevalence/risk relationships decrease with age. Although serious overprediction may occur in the young, overprediction is only moderate in the elderly. It should be stressed, however, that extensive coronary artery calcification does not necessarily indicate the presence of a significant coronary artery stenosis, and some patients with very advanced coronary calcium scores have no significant coronary artery stenosis or functional abnormality on stress perfusion or LV functional analysis.
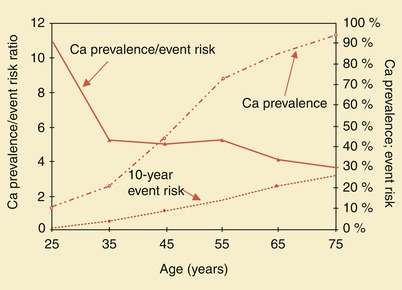
Fig. 15.5
Coronary artery calcium (Ca) prevalence, 10-year event risk, and prevalence/risk ratio in asymptomatic men. Event risk and calcium prevalence are plotted against the right axis, and prevalence/risk ratio is plotted against the left axis. The prevalence/risk curve decreases with age, showing serious overprediction in the young and moderate overprediction in the elderly
Because of high cost and radiologic exposure, we do not recommend routine screening of patients for coronary calcification. Indeed, Hoffmann and colleagues [23] have reported a multicenter trial involving 1,000 patients with suspected ACS. Compared with a standard evaluation strategy, CCTA improved the efficiency of clinical decision-making but increased downstream testing and radiation exposure with no decrease in the overall costs of care.
Variant Angina (“Prinzmetal’s Angina”)
Patients with variant angina pectoris (“Prinzmetal’s angina”) have angina at rest, often in the early morning hours, associated with ST-segment elevation on the ECG and the presence of coronary artery spasm that causes focal obliteration of a coronary artery lumen (Fig. 15.6) [24, 25].
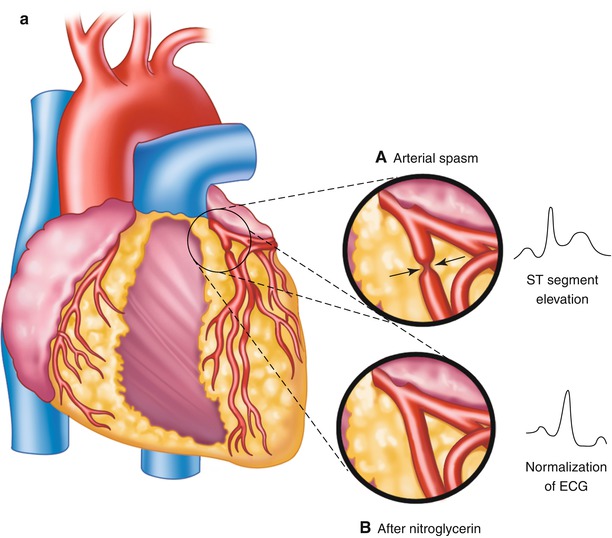
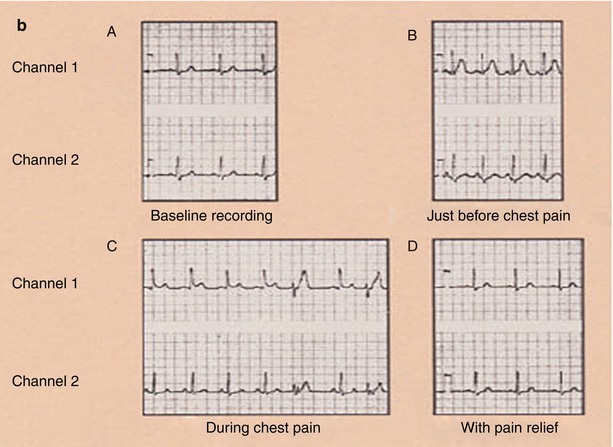
Fig. 15.6
Panel a: (Inset A) Focal obliteration of the coronary artery lumen caused by coronary arterial spasm at a spot marked by the arrows (Inset B). Panel b: The associated ST-segment elevation that occurs with coronary arterial spasm (A–D). Continuous 24-h Holter recording in a patient with coronary arterial spasm before chest pain (A, B), as chest pain begins (B), during chest pain (C), and with pain relief (D). The T-wave prominence and ST-segment elevation that occur with coronary arterial spasm after nitroglycerin administration (inset), with normalization of the electrocardiogram (ECG)
In the early descriptions of typical angina, Latham [26] and Osler [27] suggested that this entity was due to periodic spasm of a large coronary artery. Subsequently, however, clinical studies with anatomic correlations suggested that fixed atherosclerotic CAD was responsible for typical angina and MI. In 1959, Prinzmetal and coworkers [28] revived interest in coronary arterial spasm when they described a group of individuals with “variant angina.” The clinical features of this syndrome are distinctly different from those of typical angina [25, 28–55]. First, the patients described by Prinzmetal and coworkers usually had chest pain at rest rather than with physical exertion or emotional stimulation. Second, the episodes of pain tended to recur at roughly the same time(s) each day, often during the early morning hours, awakening the patient from sleep. Third, the patients with variant angina usually had ST-segment elevation on the ECG recorded during chest pain (Fig. 15.6). Fourth, the episodes of chest pain were sometimes accompanied by atrioventricular block or ventricular ectopic activity, and occasionally, the patients had transient ventricular tachycardia. Finally, the chest discomfort of variant angina was quickly relieved by nitroglycerin, after which the ST-segment elevation resolved. Prinzmetal’s patients did not undergo selective coronary arteriography, but Prinzmetal and coworkers hypothesized that patients with variant angina had severe proximal stenoses of one or more large coronary arteries in which spasm occurred periodically. Since the original description of variant angina by Prinzmetal and coworkers, many observers [28, 29, 31, 33, 37–41, 43, 47, 48] have confirmed the existence of this syndrome and have shown the presence of coronary artery spasm at sites of fixed coronary artery stenosis and in regions of the coronary vasculature where no obvious stenosis exists. In patients with clinically active coronary artery spasm, variant angina can often be induced by ergonovine maleate [30, 32, 34–36, 42, 44–46, 49, 51]. Other maneuvers that have been used to produce coronary artery spasm in susceptible patients include cold pressor testing, hyperventilation with the administration of an alkaline buffer, and the administration of methacholine (a parasympathomimetic agent), acetylcholine, or serotonin [34, 36, 49–55]. We have speculated that endothelial or adventitial injury, often in association with coronary artery stenosis, leads to the accumulation of platelets, white blood cells, mononuclear cells (including mast cells), and T cells; it also leads to the release of humoral mediators, including serotonin, thromboxane A2, prostaglandin D2, thrombin, leukotrienes, platelet activating factor (PAF), endothelin, and histamine, which singly or in combination may cause coronary artery spasm (see Figs. 15.3 and 15.6) [56–65]. The accumulation of tissue factor [66] and of local inhibitors of thrombolysis—such as the endogenous inhibitor of tissue plasminogen activator (PAI-1) [67]—and the activation of procoagulant factors, such as factors X and Xa (Fig. 15.7) [68], may also contribute to the development of thrombosis. This is especially likely to be true when endothelial injury decreases vascular concentrations of the endogenous inhibitors of inflammation, thrombosis, and vasoconstriction, i.e., nitric oxide (NO), tissue plasminogen activator, and/or prostacyclin (see Fig. 15.3) [63–65]. It also seems highly likely that the endothelium-derived vasoconstrictor, endothelin, is responsible for coronary artery constriction or spasm in some patients (Fig. 15.3) [63–65].
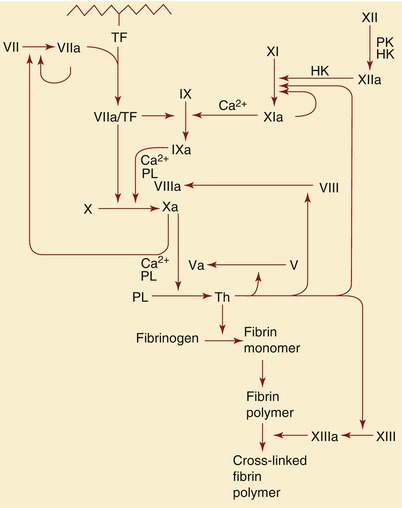
Fig. 15.7
Schematic representation of the role of tissue factor (TF) and factors IX, X, XI, XII, and VIII in the formation of thrombin (Th), as well as the subsequent role of thrombin in the formation of fibrin. HK high-molecular-weight, PK prekallikrein, PL phospholipids, PT prothrombin
Local plaque and systemic increases in catecholamine concentrations also contribute to the development of plaque thrombosis and vasoconstriction.
Unstable Angina
Angina occurring in a crescendo pattern, during limited physical activity or at rest, is known as unstable angina. The chest discomfort tends to be milder than that occurring with acute MI, being described as recurrent chest or epigastric “tightness” or “pressure” and as usually “not severe” in character. Typically, the episodes of angina last less than 30 min; they may or may not be associated with nausea. On occasion, however, unstable angina is associated with more severe and prolonged chest pain or nausea, making the clinical differentiation of unstable angina from acute MI at the bedside difficult. In these situations, serial ECGs and measurement of serum creatine kinase (CK) and its relatively specific cardiac isoenzyme, CK-MB, and of troponin I or T are needed to distinguish unstable angina from acute MI. Whereas unstable angina and non-ST-segment elevation MI (NSTEMI) both lead to ST-segment flattening or depression and T-wave flattening or inversion (Fig. 15.8), increases in troponin I or T or of CK-MB levels identify the presence of myocardial necrosis.
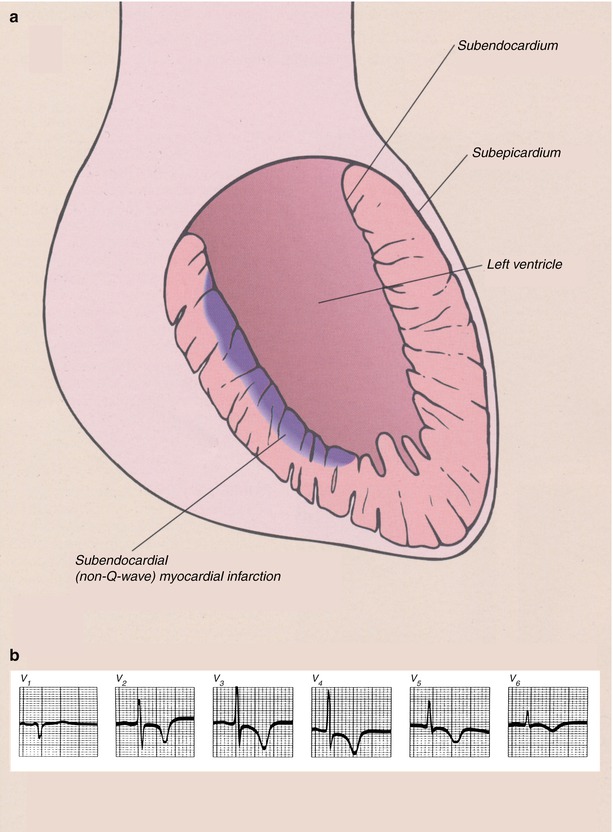
Fig. 15.8
(a) Schematic diagram showing the location of myocardial necrosis with non-ST-segment elevation myocardial infarction (NSTEMI) and a representation of the typical electrocardiographic changes. (b) Typical electrocardiogram (ECG) in a patient with acute NSTEMI. With these infarcts, the ECG is unable to provide specific evidence of the presence of the infarct, but ST-segment depression of varying magnitude and T-wave abnormalities, usually T-wave flattening or inversion, often develop. The only evolution of the electrocardiographic abnormalities is a return to the normal pattern
Unstable angina is usually caused by a primary decrease in coronary blood flow and myocardial oxygen delivery, occurring as a consequence of atherosclerotic plaque fissuring or ulceration [66–77]. The atherosclerotic plaque fissuring or ulceration is followed by platelet adhesion and aggregation at the site of plaque disruption and by transient thrombosis or subtotal coronary artery occlusion with dynamic vasoconstriction. Platelet adhesion occurs by means of platelet attachment to exposed collagen and to von Willebrand binding sites largely through platelet glycoprotein Ib receptors. Thrombosis and vasoconstriction are furthered by the local accumulation of powerful promoters of platelet aggregation and vasoconstriction at these same sites, including thromboxane A2, serotonin, adenosine diphosphate, PAF, thrombin, oxygen-derived free radicals, tissue factor, and endothelin (see Fig. 15.3) [56–65]. All of these substances but endothelin promote thrombosis. Thromboxane A2, serotonin, PAF, and thrombin are also vasoconstrictors. Endothelin is a powerful vasoconstrictor. There is also a loss of the endogenous inhibitors of thrombosis and vasoconstriction, NO, tissue-type plasminogen activator (t-PA), and prostacyclin at sites of vascular injury (Fig. 15.3) [69, 73, 75–77].
Unstable angina may also occur in individuals with a severe coronary artery stenosis or partially occluded coronary artery when myocardial oxygen demand is increased by intense emotion, tachycardia, or systemic hypertension. Alternatively, unstable angina may result from a reduction in myocardial blood flow associated with severe and progressive coronary artery atherosclerosis or dynamic coronary artery constriction associated with coronary artery spasm [24, 25].
Vulnerable, or Unstable, Atherosclerotic Plaques
Characteristics of Vulnerable Plaques
Figure 15.9 shows the characteristics of a “vulnerable,” or unstable, atherosclerotic plaque, herein defined as a plaque likely to ulcerate or fissure or otherwise to promote platelet adherence and aggregation and vasoactive mediator accumulation leading to the development of thrombosis, dynamic vasoconstriction, and fibroproliferation. The morphologic characteristics of a vulnerable plaque include a thin fibrous cap, the presence of inflammatory cells on or beneath the atherosclerotic plaque surface, and an adjacent lipid core. Casscells and associates [78] showed that such plaques in the human carotid artery have temperature heterogeneity, with temperatures that vary by 0.2–2 °C in the part of the plaque where the inflammation exists (Fig. 15.10). More recently, others have confirmed these observations and shown the same temperature heterogeneity in vivo in human coronary arteries in patients with unstable angina and acute MIs (Fig. 15.11) [79]. In vivo studies have shown that the temperature heterogeneity is more commonly in the 0.15–1 °C range in the vulnerable atherosclerotic plaque. Buffon, Pasterkamp, and others [80–82] have suggested that unstable atherosclerotic plaques may develop as part of widespread inflammation and that a substantial number of patients have multiple vulnerable plaques simultaneously.
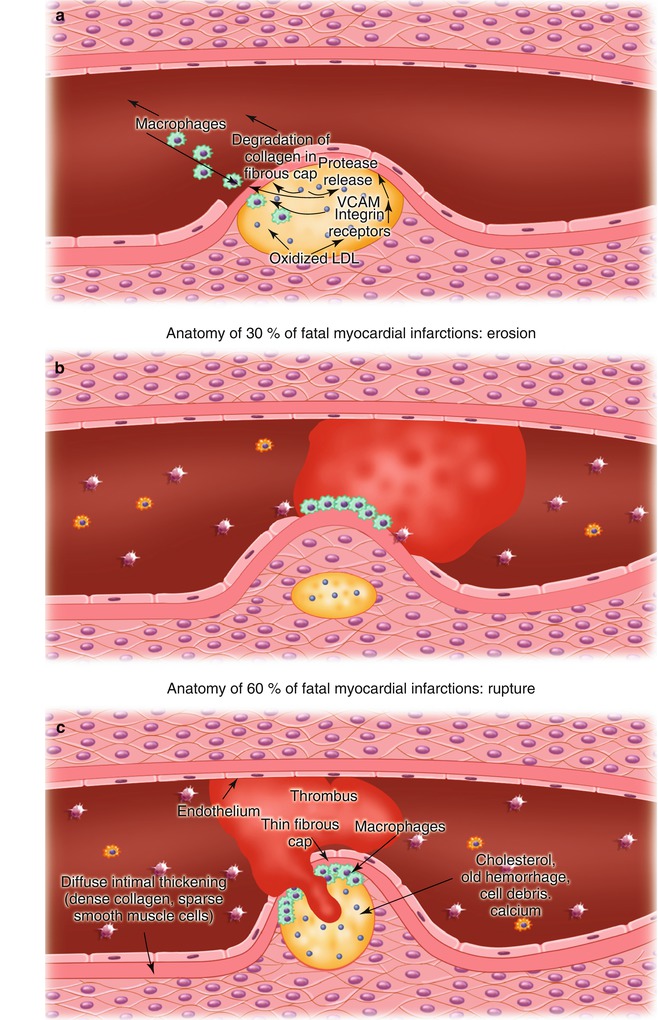
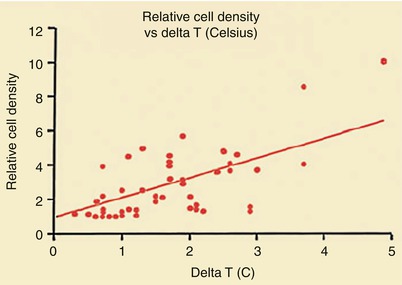
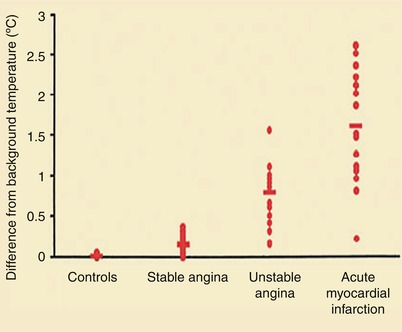
Fig. 15.9
(a) Potential mechanisms responsible for atherosclerotic plaque fissuring and ulceration. Oxidized low-density lipoprotein (LDL) within the atherosclerotic plaque promotes the upregulation of vascular cell adhesion molecules (VCAM) and other integrins, resulting in the recruitment of inflammatory cells, primarily monocyte-derived macrophages, but including activated T cells and mast cells; subsequent release of proteases from the mononuclear cells; and degradation of collagen in the fibrous cap, leading to its fissuring and ulceration. (b) Atherosclerotic plaque fissuring, leading to platelet adhesion and aggregation and thrombosis. (c) Atherosclerotic plaque ulceration and thrombosis
Fig. 15.10
Relationship between temperature heterogeneity and inflammatory cell presence in human carotid plaques removed at carotid endarterectomy
Fig. 15.11
Increased temperature of infarct-related plaques in patients with stable angina, unstable angina, and acute MI, compared with control patients (From Stefanadis et al. [79]. Reprinted with permission from Wolters Kluwer Health)
Vulnerable Plaques and Increased Risk of Future Adverse Events
Several studies have shown that patients who have unstable angina, NSTEMI, and ST-segment elevation MI (STEMI) with elevated serum C-reactive protein (CRP) levels at hospital discharge, elevations in serum troponin I or T levels at hospital admission, or increases in serum interleukin-6 concentrations or of multiple biomarkers at hospital admission have an increased risk of future coronary events, presumably reflecting the importance of inflammation in the instability of unstable atherosclerotic plaques (Figs. 15.12, 15.13, 15.14, 15.15, 15.16 and 15.17) [83–94]. Increases in troponin I and T levels identify myocardial necrosis and possibly more extensive plaque instability, longer duration of platelet-derived thrombosis and vasoconstriction, and sometimes platelet emboli and occlusive disease of distal arteries (Figs. 15.13 and 15.14) [83, 85–88, 90, 91, 93, 94]. Table 15.2 lists other biomarkers that have been shown to be predictive of future cardiovascular events (Figs. 15.15 and 15.16) [95–103].
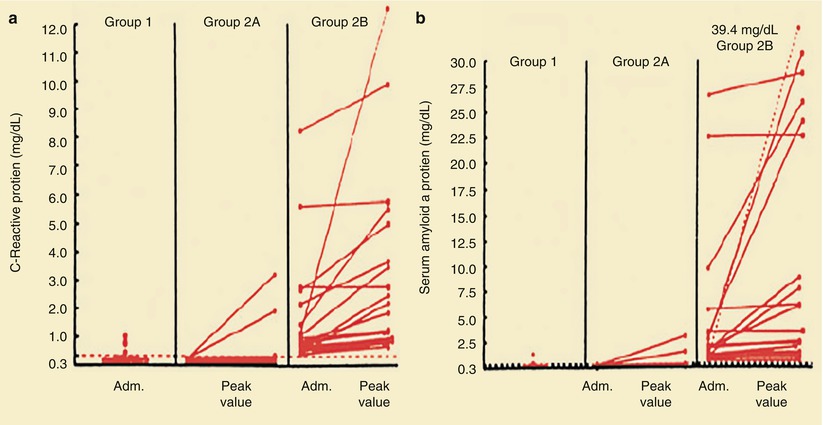
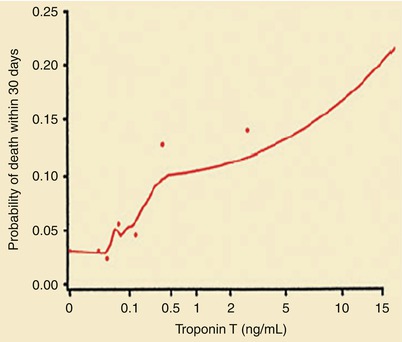
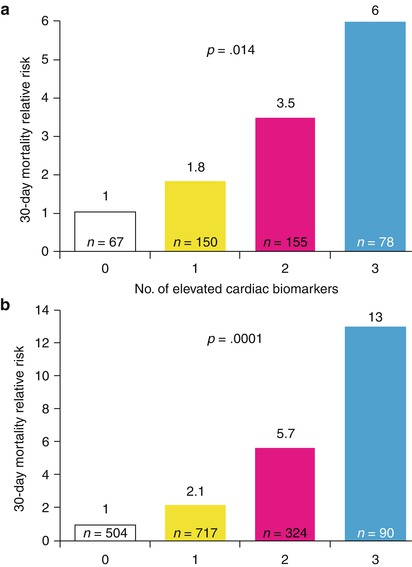
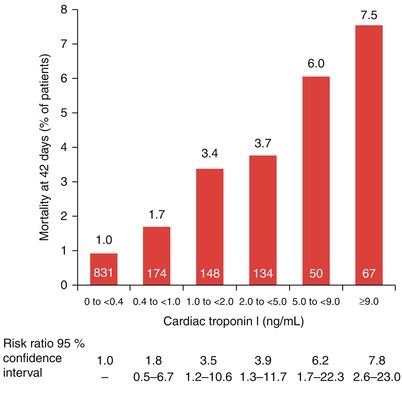
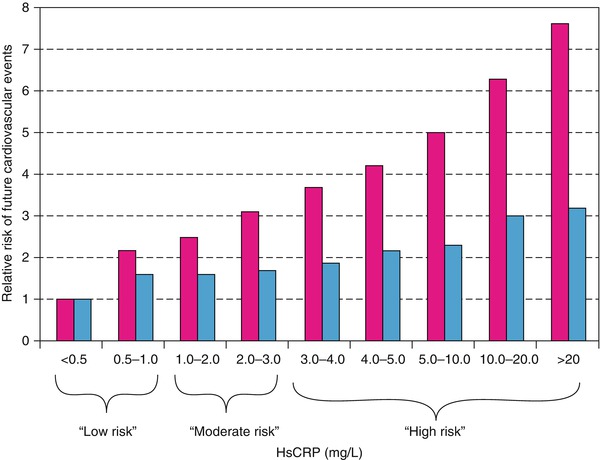
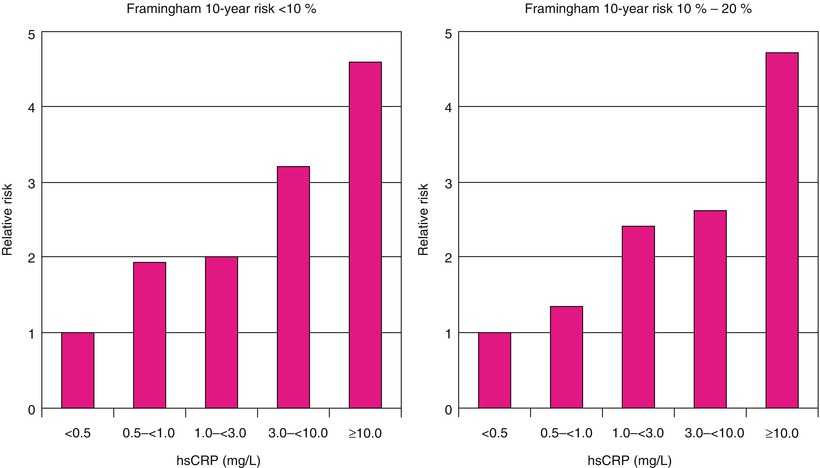
Fig. 15.12
Plasma levels of C-reactive protein (a) and serum amyloid A protein (b) in patients with stable angina (group 1), patients with unstable angina and levels of C-reactive protein <0.3 mg/dL on admission (Adm.) (Group 2A), and patients with unstable angina and levels of C-reactive protein ≥0.3 mg/dL on admission (Group 2B). Circles denote urgent coronary artery bypass or angioplasty; squares, cardiac death or MI. In one patient, the peak value for serum amyloid A protein, which exceeded the values on the scale, is shown numerically and joined by a dashed line to the corresponding value on admission. The level of C-reactive protein is <0.3 mg/dL (horizontal line in a) in 90 % of normal subjects. The median normal level of serum amyloid A protein is 0.3 mg/dL (dashed horizontal line in b); 96 % of normal subjects have levels of serum amyloid A protein <1.0 mg/dL (From Liuzzo et al. [83]. Reprinted with permission from Massachusetts Medical Society)
Fig. 15.13
Probability of death within 30 days according to the serum troponin T level at hospital admission. The dots represent simple estimates of mortality derived from ranges of the troponin T level that included at least 70 patients (From Ohman et al. [84]. Reprinted with permission from Massachusetts Medical Society)
Fig. 15.14
Relative 30-day mortality risks in OPUS-TIMI 16 (a) and TACTICS-TIMI 18 (b) in patients stratified by the number of elevated cardiac biomarkers
Fig. 15.15
The mortality rate at 42 days for patients admitted to the hospital with unstable angina or a non-ST-segment elevation myocardial infarction (NSTEMI) (non-Q wave MI) based on their initial cardiac troponin serum levels. The number of patients in each category is shown within each red bar (From Antman et al. [85]. Reprinted with permission from Massachusetts Medical Society)
Fig. 15.16
Relative risks of future cardiovascular events across a full clinical range of high-sensitivity C-reactive protein (HsCRP) values. Red bars represent crude relative risks; blue bars, risk adjusted for Framingham Risk Score
Fig. 15.17
Relative risks of future cardiovascular events among those with calculated 10-year Framingham risks <10 % (left) and between 10 and 20 % (right). hsCRP, high-sensitivity C-reactive protein
Table 15.2
Biomarkers to predict the prognosis in patients with unstable angina and non-ST-segment elevation myocardial infarction (NSTEMI)
C-reactive protein |
Brain natriuretic peptide (BNP) |
CD40L and CD40 |
Myeloperoxidase |
Pregnancy-associated protein |
Serum amyloid protein |
Vascular cell adhesion molecule (VCAM) |
Intercellular adhesion molecule (ICAM) |
Interleukin-6 |
Asymmetric dimethylarginine |
Troponins |
Phospholipase A2 |
Sabatine and colleagues [104] have also shown that elevations in serum troponin, CRP, and brain natriuretic peptide (BNP) levels each provide unique prognostic information in patients with ACS. A relatively simple multimarker strategy that categorizes patients with ACS based on the number of elevated biomarkers at admission allows risk stratification over a range of short- and long-term major cardiac events (Fig. 15.14) [104].
Berk and coworkers [86] were among the first to demonstrate an increase in CRP in patients with acute coronary heart disease syndromes. Subsequently, several studies, including those by Liuzzo and colleagues [83], revealed a poorer prognosis for patients with unstable angina who had increased serum CRP and serum amyloid-A protein values. Ridker and associates [94] (Fig. 15.17) and Koenig and coworkers [90] demonstrated that increases in serum CRP predict the development of future coronary events, even in otherwise apparently healthy individuals. Figure 15.12 shows that for patients with unstable angina and NSTEMI with CRP values of 0.3 mg/dL or higher, there is an increased risk of future coronary events, including urgent coronary artery bypass surgery or angioplasty, cardiac death, or MI. Similar data have been provided for patients who have elevated serum fibrinogen values at hospital discharge [91].
Several groups have shown the importance of increases in serum levels of troponin I as prognostic factors indicative of increased future risk for patients with ACS. In a multicenter study of 1,404 symptomatic patients, Antman and colleagues [85] found a relation between mortality at 42 days and the serum cardiac troponin I levels at patient admission to the hospital (Fig. 15.15). In the 573 patients with cardiac troponin I levels of 0.4 ng/mL and greater, the mortality rate at 42 days was significantly higher than in the 831 patients with cardiac troponin I levels of less than 0.4 ng/mL. For each increase of 1 ng/mL in the cardiac troponin I level, there was an associated significant increase in the risk ratio for death, after adjustment for baseline characteristics that were independently predictive of mortality, including ST-segment depression and age 65 years or older. Similar data have been provided for serum values of troponin T in patients with unstable coronary heart disease. Lindahl and associates [92] found that the risk of cardiac events in these patients increased with rising maximal levels of troponin T obtained in the initial 24 h after admission. The lowest quartile (<0.06 μg/L) constituted a low-risk group, the second quartile (0.06–0.18 μg/L) an intermediate group, and the third highest quartile (≥0.18 μg/L) a relatively high-risk group with a 4.3, 10.5, and 16 % risk of MI, cardiac death, or both, respectively. Biasucci and coworkers [87] demonstrated an increased risk for future coronary events in patients whose serum interleukin-6 level increased during hospital admission. Other serum markers that, when elevated, are associated with future vascular and myocardial events include CD40L, CD40, serum amyloid protein (SAP), pregnancy-associated protein, vascular cell adhesion molecules (VCAMs), intracellular adhesion molecules (ICAMs), and BNP (Table 15.2) [89].
Matrix Metalloproteinases and Their Tissue Inhibitors
Stability of an atherosclerotic plaque depends largely on the structural integrity of its fibrous cap, which is composed primarily of extracellular matrix components rich in collagen. Available evidence suggests that in the catabolism of the structural macromolecules, the release of matrix-degrading enzymes—that is, matrix metalloproteinases (MMPs)—causes a dissolution of the fibrous cap of the plaque [105–110]. The MMP family includes at least 19 structurally related, zinc-dependent enzymes that function at physiologic pH in the extracellular space. In addition, researchers have identified membrane-type (MT) MMPs that have the designations MT1-MMP through MT4-MMP.
The MMPs are broadly classified into three main groups on the basis of substrate specificity. They include the collagenases (MMP-1, −8, and −13), which degrade intact fibrillar collagens. The gelatinases (MMP-2 and −9) hydrolyze the denatured collagen fibril and basement membrane collagen type IV. The stromelysins (MMP-3, −7, −10, and −11) have broad substrate specificity. For some of the remaining MMPs, substrate specificity has not yet been identified.
Regulation of MMPs occurs at three levels: (1) control of the rate of gene transcription; (2) conversion of the inactive translational product, and inactive zymogen precursor, to the active form; and (3) inactivation by a family of endogenous inhibitors known as tissue inhibitors of metalloproteinases (TIMPs).
Human peripheral blood monocytes produce little MMP, but differentiation into macrophages yields higher levels of MMP-9 [105–110]. Secretion of MMP-9 from macrophages can be induced by tumor necrosis factor-α, interleukin-1β, and CD40. Expression of collagenase (MMP-1) and stromelysin (MMP-3) in macrophages is regulated by certain bacterial products, including lipopolysaccharide (endotoxin) and yeast zymosan. Activated T lymphocytes may cause collagenase and stromelysin expression in human monocyte-derived macrophages.
The MMPs are secreted from cells as inactive zymogens and require activation in the extracellular milieu before they are capable of degrading extracellular matrix molecules. Macrophage-derived reactive oxygen species activate MMP-2 and MMP-9, and thrombin activates MMP-2. Once processed from the zymogen to active form, MMPs themselves can trigger activation of other members of the MMP family. Mast cells within atheroma may release serine proteinases that activate MMPs.
The endogenous inhibitors known as TIMPs help regulate MMP activity under normal circumstances. They exhibit sequence homology and share domains of identical protein structures that consist of highly conserved N-terminal regions believed to be critical for inhibition of the enzyme.
The balance between MMP activities and their inhibition by TIMPs is important in the maintenance of homeostasis for the extracellular matrix. Excessive MMP activity occurs in a number of disease states, including metastatic cancer, rheumatoid arthritis, and glomerulosclerosis. There is evidence that MMPs play a role in accelerated connective tissue turnover associated with degenerative diseases of vessels, including aortic aneurysms and vein graft stenoses, as well as migration of smooth muscle cells from the media to the intima after arterial injury.
Several of the MMPs and TIMPs have been found in atherosclerotic plaques [105–110], including MMP-1 (collagenase), MMP-9 (gelatinase B), and stromelysin-1 (MMP-3). Each of these has been found in the fibrous cap, the atherosclerotic lesions’ shoulders, and the base of the lipid core. MMP-7 (matrilysin) is found primarily in macrophages overlying the lipid core. A high expression of MMPs has also been detected in lipid-laden macrophages in experimental animal lesions.
Similarly, constitutive expression of TIMP-1 and −2 has been described in both normal and diseased arteries, and TIMP-3 has been found in atheromas. Thus, the spatial distribution of selected TIMPs within atheromas correlates with that described for MMP expression. Most of the MMPs and TIMPs in atheromas are in macrophages, thereby identifying the macrophage as most likely a key participant in the regulation of the balance between synthesis and degradation of extracellular matrix macromolecules in the atherosclerotic plaque.
Available evidence suggests a probable role for metalloproteinase release in excess of TIMP presence, leading to degradation of collagen in the fibrous cap and the subsequent fissuring and ulceration that predisposes to unstable angina and acute MI (see Fig. 15.9). The proposed scheme includes the oxidation of low-density lipoprotein (LDL) within the plaque; the chemoattractant influence of oxidized LDL and other oxidation products that promote the expression of selected adhesion molecules, including VCAMs and ICAMs; and the subsequent recruitment of monocyte-derived macrophages, activated T lymphocytes, and mast cells within the plaque. The release of selected MMPs in excess of their TIMP concentrations locally degrades plaque collagen within the fibrous cap and leads to fissuring and ulceration of the atherosclerotic plaque. Future clinical trials involving inhibitors of selected MMPs are anticipated, but the redundancy within the system—that is, the number of MMPs and other proteases within plaques that are nonmetalloenzymes—may make effective inhibition of these plaque-degrading enzymes difficult. Other approaches to the reduction of inflammation and the recruitment of monocyte-derived macrophages that may be protective of atherosclerotic plaques include inhibition of macrophage homing to unstable atherosclerotic plaques by using inhibitors of VCAM and ICAM [111] or of antioxidants, inhibitors of selected cytokines (especially tumor necrosis factor-α), NO donors, and inhibitors of nuclear factor κB (NF-κB). Marked lipid lowering may also be achieved with medications capable of providing reductions in serum cholesterol and LDL, most especially statins [112–114]. Although current guidelines recommend the evaluation of fasting lipid profiles and use of statin therapy in all ACS patients, many high-risk patients who could most benefit from these interventions do not receive them [112].
Direct Evaluation of Vulnerable Plaques
Because the risk of coronary occlusion may depend more on the nature of atherosclerotic plaques than on the degree of luminal narrowing, there is a need for imaging techniques that can directly evaluate vulnerable plaques and identify those most likely to rupture. Traditional coronary angiography is unable to achieve this goal. Figure 15.18 summarizes the methods that are currently available for direct evaluation of plaques [115]. CCTA, MRI, and nuclear imaging techniques have limited usefulness for this purpose, but combinations of these techniques may expand their usefulness in the future. Although numerous other modalities are being investigated for plaque evaluation, most of them are regarded as experimental. A current major focus of research is the development of nanotechnology CT contrast agents, such as gold or bismuth nanoparticles, which would allow targeted molecular imaging [17].
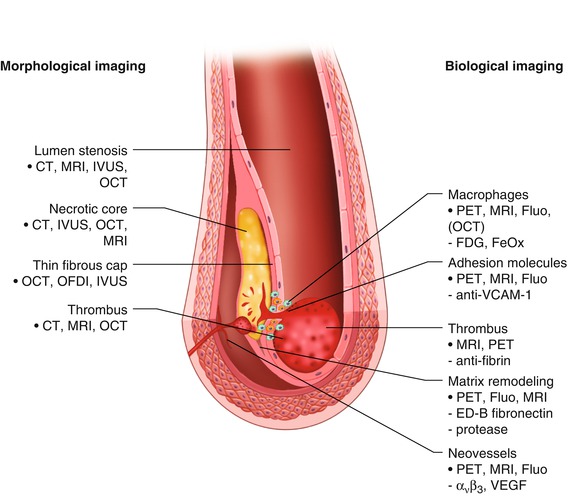
Fig. 15.18
Depicted are two tools for visualizing vulnerable plaques: morphological and biological imaging
One of the few approaches to direct plaque evaluation already being used in the clinical setting is intravascular ultrasonography (IVUS). By providing real-time, high-resolution, cross-sectional images from within blood vessels, IVUS can both identify plaque and assess its quality [115, 116]. Conventional gray-scale imaging is limited for this purpose, but virtual histology (VH) techniques can assess plaque components by spectrally analyzing IVUS backscatter radiofrequency signals; the results are color coded and represented as tissue maps (Fig. 15.19) [115]. The recent PROSPECT trial [118], which prospectively tracked the natural history of unstable plaques, provided clinical evidence that IVUS and VH-IVUS are able to determine the differences between high-risk and low-risk lesions (Fig. 15.20) [117].
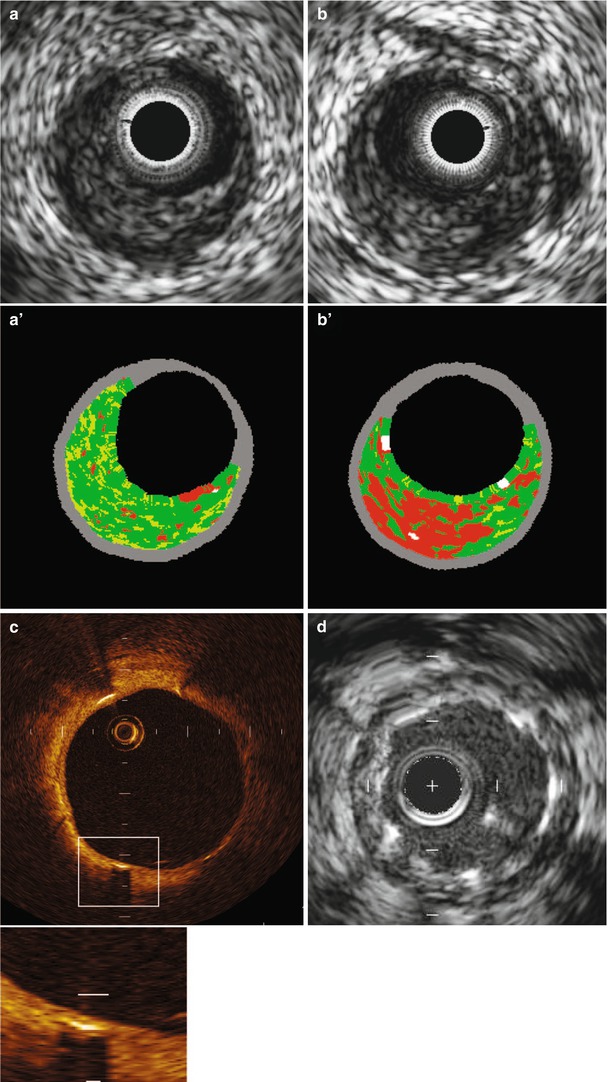
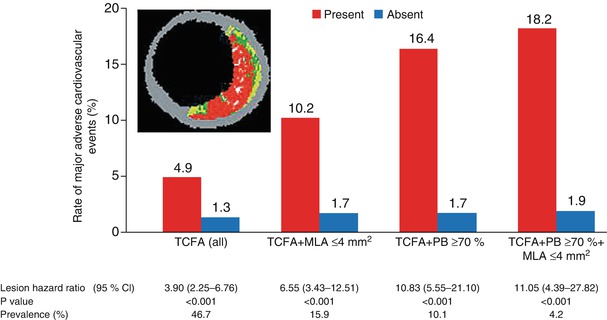
Fig. 15.19
(a) Grayscale and VH (a’) image that is classified as pathological intimal thickening. (b) Grayscale and VH (b’) image that is classified as thick-cap fibroatheroma. Although both grayscale IVUS images are quite similar, VH images show different morphology. An OCT image (c) of a chronically implanted stent shows clear stent coverage not seen in IVUS (d) (From Maehara et al. [117]. Reprinted with permission from Wolters Kluwer Health)
Fig. 15.20
Event rates for lesions that were and those that were not thin-cap fibroatheromas, at a median follow-up of 3.4 years. Event rates associated with 595 nonculprit lesions that were characterized as thin-cap fibroatheromas (TCFA) and 2,114 that were not by means of radiofrequency intravascular ultrasonographic imaging are shown according to minimal luminal area (MLA) and plaque burden (PB) as detected on gray-scale intravascular ultrasonography. The inset shows an example of a thin-cap fibroatheroma imaged by radiofrequency ultrasonography. Data on prevalence are for one or more such lesions per patient. Lesions in patients with indeterminate events were excluded. CI denotes confidence interval (From Stone et al. [118]. Reprinted with permission from Massachusetts Medical Society. For additional details, see Table 6 in the Supplementary Appendix of that article [118])
Another emerging option for the intravascular evaluation of vulnerable plaques is optical coherence tomography (OCT), which is an optical analogue to IVUS but has much better resolution [115]. Thus, OCT is not only highly sensitive and specific for detecting lipid and calcified elements but also an extremely promising tool for the study of plaque components [117] (Fig. 15.19). Its limitations include a fairly shallow depth of tissue penetration and interference from intracoronary blood. Nevertheless, OCT appears to be the most promising modality for imaging plaque morphology [116]. In particular, OCT allows an estimate of atherosclerotic cap thickness, which itself is a predictor of fissuring or rupture of an atherosclerotic plaque. The recent work of Virmani and associates has provided valuable insights into the importance of fibrous cap thickness in plaque stability [119, 120].
Acute Myocardial Infarction
An acute MI (Table 15.3 [121]) occurs when coronary blood flow and myocardial oxygen delivery are severely reduced for more than 20 min. The infarct begins on the inner wall or subendocardium of the heart and is confined there in the first 30 min to 2 h (Fig. 15.8; non-Q wave or NSTEMI). These lesions are generally subendocardial infarcts. If the coronary artery thrombosis is transient or does not cause complete coronary artery occlusion, the infarct usually remains largely confined to the subendocardium (or mid-wall of the heart) and a non-Q wave infarct or NSTEMI develops. However, if the coronary artery occlusion is sustained for longer periods, the myocardial necrosis progresses vertically outward toward the epicardium in the ensuing 2–3 h (“Q-wave” infarct or STEMI) (Fig. 15.21).
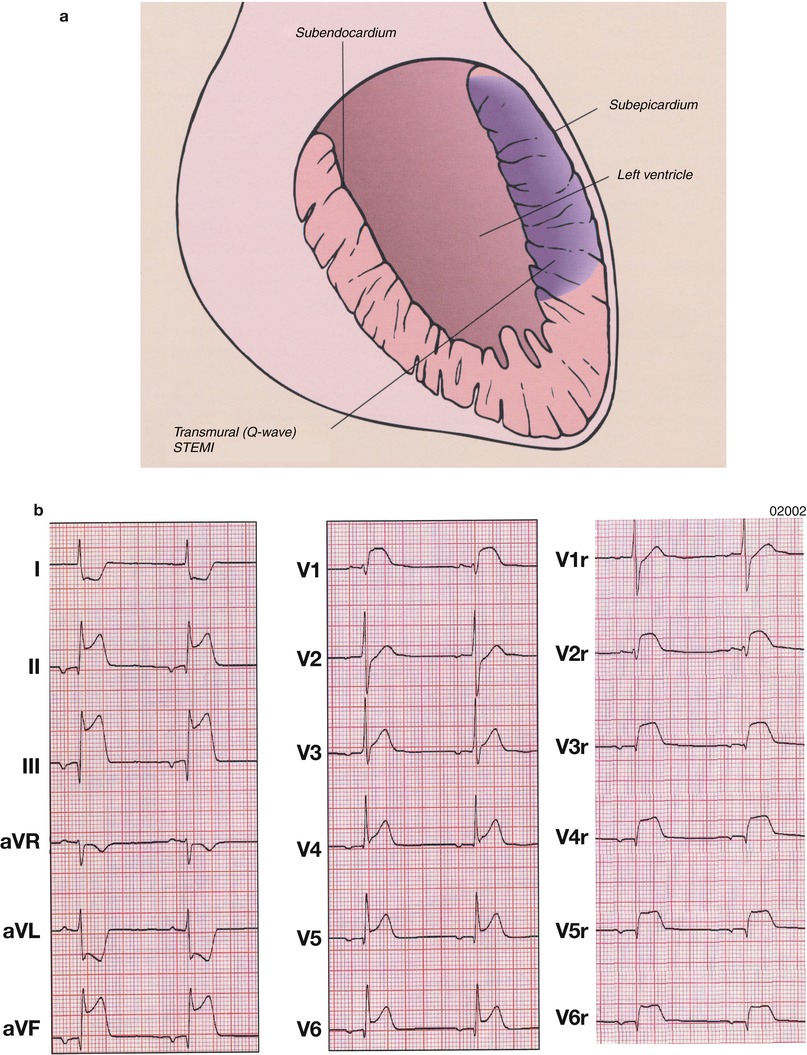
Table 15.3
Definition of myocardial infarction
Criteria for acute myocardial infarction |
The term acute myocardial infarction (MI) should be used when there is evidence of myocardial necrosis in a clinical setting consistent with acute myocardial ischemia. Under these conditions any one of the following criteria meets the diagnosis for MI: |
Detection of a rise and/or fall of cardiac biomarker values [preferably cardiac troponin (cTn)] with at least one value above the 99th percentile upper reference limit (URL) and with at least one of the following: |
Symptoms of ischemia. |
New or presumed new significant ST-segment–T wave (ST–T) changes or new left bundle branch block (LBBB). |
Development of pathological Q waves in the ECG. |
Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality. |
Identification of an intracoronary thrombus by angiography or autopsy. |
Cardiac death with symptoms suggestive of myocardial ischemia and presumed new ischemic ECG changes or new LBBB, but death occurred before cardiac biomarkers were obtained, or before cardiac biomarker values would be increased. |
Percutaneous coronary intervention (PCI) related MI is arbitrarily defined by elevation of cTn values (>5 × 99th percentile URL) in patients with normal baseline values (≤99th percentile URL) or a rise of cTn values >20 % if the baseline values are elevated and are stable or falling. In addition, either (i) symptoms suggestive of myocardial ischemia or (ii) new ischemic ECG changes or (iii) angiographic findings consistent with a procedural complication or (iv) imaging demonstration of new loss of viable myocardium or new regional wall motion abnormality are required. |
Stent thrombosis associated with MI when detected by coronary angiography or autopsy in the setting of myocardial ischemia and with a rise and/or fall of cardiac biomarker values with at least one value above the 99th percentile URL. |
Coronary artery bypass grafting (CABG) related MI is arbitrarily defined by elevation of cardiac biomarker values (>10 × 99th percentile URL) in patients with normal baseline cTn values (≤99th percentile URL). In addition, either (i) new pathological Q waves or new LBBB, or (ii) angiographic documented new graft or new native coronary artery occlusion, or (iii) imaging evidence of new loss of viable myocardium or new regional wall motion abnormality. |
Criteria for prior myocardial infarction |
Any one of the following criteria meets the diagnosis for prior MI: |
Pathological Q waves with or without symptoms in the absence of non-ischemic causes. |
Imaging evidence of a region of loss of viable myocardium that is thinned and fails to contract, in the absence of a non-ischemic cause. |
Pathological findings of a prior MI. |
Fig. 15.21
(a) Schematic diagram of the topographic location of myocardial necrosis with ST-segment-elevation myocardial infarction (STEMI) and the associated electrocardiographic changes. (b) Atrial rhythm, rate 50 beats/min. Q waves in leads II, III, aVF and V6 indicate a conduction delay in the inferolateral wall and ST-segment elevation in leads II, III, aVF and 4–6, consistent with an inferolateral infarction and right ventricular involvement. The right precordial leads show prominent ST-segment elevation V2r–V6r. Note also ST-segment depression in leads aVR and aVL, which is typical of an acute inferior wall infarction
Herrick [2] described acute MI caused by coronary artery thrombosis in 1912. Subsequently, the role of coronary artery thrombosis in causing MI was debated [92] until DeWood and colleagues [122] demonstrated by means of coronary arteriography that coronary artery thrombosis is virtually always the cause of acute Q-wave infarcts or STEMIs. By documenting detailed clinicopathologic correlations, Buja and Willerson [70] confirmed the association between thrombosis of the infarct-related coronary artery and the development of acute Q-wave infarction or STEMI. Ninety percent or more of STEMIs involve prolonged occlusive coronary artery thrombosis in the infarct-related coronary artery [70, 122, 123].
As noted above, STEMIs are usually caused by persistent thrombotic occlusion of a coronary artery, resulting in sustained reductions in coronary blood flow and myocardial oxygen availability. Occasionally, increases in myocardial oxygen demand above the ability of a stenotic coronary artery to delivery oxygen will cause an MI, often NSTEMI. Such increases in oxygen demand occur in some patients with CAD who have severe systemic arterial hypertension, sustained tachycardia, or both. Alternatively, sustained reductions in myocardial oxygen delivery associated with severe systemic arterial hypotension may lead to MI, again usually NSTEMI. Approximately 30 % of patients with NSTEMI have an occlusive thrombus in the infarct-related artery [70, 124, 125]. Nevertheless, most NSTEMI patients have transient coronary artery occlusion that is initiated by platelet aggregation and involves associated vasoconstriction probably lasting more than 30 min and up to 2 h (see Figs. 15.3 and 15.8).
Local accumulation of endothelin causes marked vasoconstriction. Endothelin, serotonin, adenosine diphosphate, and thrombin are mitogens. They likely contribute to subsequent local fibroproliferation with increases in the neointima, further narrowing the lumen of the endothelium-injured coronary artery [126]. In 30 % (or more) of patients with unstable angina and NSTEMI, there is a rapid anatomic progression in the severity of the coronary luminal diameter narrowing, most likely associated with the inclusion of organized thrombus within the plaque and the fibroproliferation that follows plaque fissuring and ulceration [126]. Reductions in fibrinolytic capability at sites of vascular endothelial injury—associated with decreases in vascular tissue concentrations of prostacyclin, tissue plasminogen activating factor, and NO—undoubtedly contribute to coronary artery thrombosis, vasoconstriction, and fibroproliferation at these same sites (see Fig. 15.3) [42, 44, 46].
We have suggested that unstable angina, NSTEMI, and STEMI represent a continuum pathophysiologically [63–65]. The process begins with a coronary endothelial injury, usually atherosclerotic plaque ulceration or fissuring. The degree of coronary artery stenosis where plaque ulceration or fissuring occurs may be mild or severe. Approximately half of the coronary stenoses where plaque fissuring or ulceration occurs are sites of less than 50 % luminal diameter narrowing [127]. We have suggested that when platelet-fibrin thrombus and associated severe vasoconstriction persist for less than 20 min and often recur, the clinical syndrome of unstable angina develops [63–65]. However, when the reduction in coronary blood flow and oxygen delivery to the heart is more prolonged, lasting 30 min to 1–2 h, NSTEMI occurs. When the period of inadequate myocardial oxygen delivery persists for more than 2 h, STEMI results (see Fig. 15.3) [63–65]. If unstable angina and acute MI are viewed in this manner, it is easy to appreciate that patients with unstable angina and NSTEMI have “aborted” STEMIs and therefore, remain at risk for a new infarction and its consequences in the ensuing 6 weeks [70, 125, 128–131]. The risk for renewed unstable angina and MI persists until the endothelial injury is repaired. Other causes of endothelial injury may also lead to this same sequence of events, including endothelial injury associated with systemic arterial hypertension, flow shear stress, smoking, diabetes mellitus, infection, aging, immune complex deposition, substance abuse (e.g., cocaine), and the placement of a coronary artery catheter into a coronary artery, especially during percutaneous transluminal coronary angioplasty (PTCA) and stent insertion [63–65].
Fissuring and ulceration of a plaque often (but not always) occur in the plaque’s asymmetric portion, or “shoulder region,” and involve a thin fibrous cap that is lipid laden. Inflammation at sites of thin fibrous plaques with adjacent lipid cores is the best predictor of a vulnerable atherosclerotic plaque, i.e., one likely to fissure or ulcerate [66–69, 71, 73–76]. Inflammation is characterized in the unstable plaque by the accumulation of monocyte-derived macrophages, activated T cells, and mast cells. Most likely, proteases released from the infiltrating mononuclear cells contribute to thinning of the fibrous cap through the degradation of collagen and subsequent atherosclerotic plaque fissuring and ulceration (Fig. 15.9) [64, 65, 74, 105–110, 126–138].
Physical Examination/Bedside Findings
The patient with unstable angina and NSTEMI and STEMI typically appears distressed. There may be no localizable finding, or the patient may have an audible S4, cardiac enlargement, congestive heart failure (CHF), acute mitral insufficiency, an acute ventricular septal defect (VSD), or evidence of peripheral vascular disease.
Between episodes of unstable angina, the ECG at rest may be normal or may show the same ST-T wave changes or previous MI found in patients with stable angina. However, often the ECG shows ST-T wave changes during angina—usually ST-segment depression or T-wave flattening or inversion in patients with unstable angina and NSTEMI—reflecting alterations in myocardial perfusion of the “culprit” artery. Similar ECG changes occur in patients with unstable angina and NSTEMI, making unstable angina and NSTEMI impossible to differentiate on the basis of the ECG alone. On occasion, episodes of unstable angina and the development of NSTEMI are not associated with ST-T wave changes on the ECG, but this is unusual.
Echocardiographic findings are highly variable, but they may include reversible or fixed reductions in regional wall motion or abnormalities in global LV function, depending on the reversibility of ischemia, the presence of an MI, and its size. In some patients, reductions in regional wall motion occur with episodes of rest angina and resolve as the angina resolves.
The chest radiograph also shows great variability, ranging from a normal-sized heart to cardiomegaly with CHF. Hemodynamic findings are similar to those observed during angina in patients with stable angina, that is, one expects to see increases in the mean pulmonary capillary wedge and LV end-diastolic pressures that are dynamic and that resolve as the angina disappears. Patients with angina may also develop transient mitral insufficiency secondary to mitral valve papillary muscle dysfunction.
Coronary artery spasm leads to abrupt and dynamic decreases in myocardial oxygen delivery (Fig. 15.6) [24, 25]. With a primary decrease in coronary blood flow, there is no association between the development of angina and exertion, and the majority of anginal episodes occur during limited activity and at rest. These patients usually have little or no change in heart rate or blood pressure before the onset of pain. The pain occurs first and may be followed by increased blood pressure and/or heart rate.
In patients with unstable angina, continuous electrocardiographic monitoring often documents a transient ST-segment change immediately before the onset of chest pain. This change may be ST-segment elevation indicating transmural ischemia as a consequence of vasoconstriction in a major epicardial coronary artery and/or platelet-initiated transient coronary artery thrombosis (see Figs. 15.3 and 15.6) with ST-segment depression [139–141]. Most commonly, the patient with unstable angina has ST-segment depression when subendocardial ischemia develops. In some individuals, ST-segment alterations occur in the absence of chest pain. This so-called silent ischemia [141] has the same prognosis as painful episodes of ischemia [139–141].
Clinical Recognition
History
The medical history is of utmost importance in the recognition of an acute MI [1, 2, 142]. Typically, the patient complains of severe chest or epigastric pain that persists until analgesic medication is administered and/or the obstructed coronary artery is opened by means of percutaneous coronary intervention (PCI) with stent placement. The pain usually lasts 30 min or longer. It often is described as being retrosternal or left precordial—a heaviness, tightness, or “weight on my chest”—and associated with nausea and diaphoresis. The pain may radiate to the back, neck, jaw, or left arm, particularly down that arm’s ulnar aspect. Occasionally, only the back, jaw, left arm, or neck is affected. The pain is usually described as the most severe the individual has ever experienced. It gradually builds in severity, reaches a peak, and then recedes. Some patients with acute MIs have unstable angina for hours to days before their infarcts. It should be emphasized, however, that 10–20 % of patients with diabetes mellitus who have MIs have “silent” (i.e., painless) ones [143–145]. Silent MIs also occur in patients who develop CAD (transplant vasculopathy) after cardiac transplantation, occasionally in individuals with neuromyopathic abnormalities, and in some seemingly otherwise normal subjects. One should have a high index of suspicion if a patient presents with new CHF, ventricular arrhythmias, hypotension, a heart murmur of mitral insufficiency, or systemic embolic events. A silent MI also should be suspected if the patient is resuscitated from sudden death, especially when diabetes mellitus is present. Serial serum measurements of CK, CK-MB, and troponin I or T levels and serial ECGs should be obtained in such patients.
Physical Examination
Patients with small MIs, particularly NSTEMIs, often have no detectable abnormality on physical examination. At the other extreme, patients with extensive damage to the left ventricle, usually anterior STEMIs with ≥40 % irreversible injury of the LV mass, often develop “power-failure” complications of their infarcts, including cardiogenic shock, severe LV failure, or medically refractory arrhythmias (Fig. 15.22) [146–150].
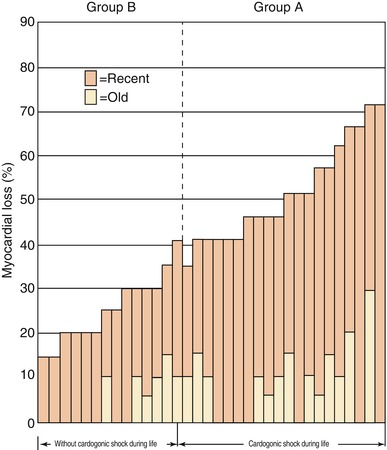
Fig. 15.22
“Power-failure” complications of MI, including cardiogenic shock, develop in patients with the most extensive myocardial necrosis from their infarcts (From Page et al. [146]. Reprinted with permission from Massachusetts Medical Society)
Inspection and Palpation
On physical examination of patients with an acute MI, the findings depend primarily on the extent of the myocardial damage. Most patients are diaphoretic and in obvious discomfort. Those with extensive myocardial damage have a reduction in systemic arterial blood pressure, ranging from mild to severe, including the development of cardiogenic shock. Cardiogenic shock is defined as hypotension resulting from extensive myocardial damage. The reduced blood pressure is inadequate for normal systemic perfusion, so cool skin, mental confusion, and oliguria are usually present. Patients with extensive LV myocardial necrosis may also have an alternating pulse force (pulsus alternans) (Fig. 15.23) and frequent premature ventricular beats.
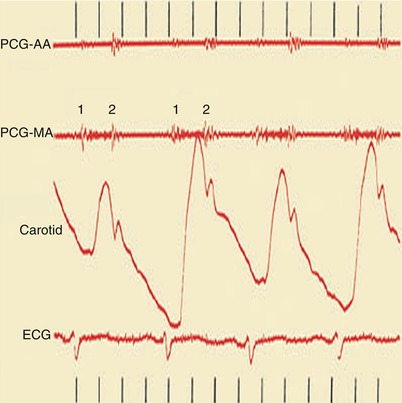
Fig. 15.23
Heart sounds (top), carotid pulse tracing (middle), and electrocardiogram (ECG) (bottom). Pulsus alternans is shown as the alternating height in the pulse wave tracing in a woman with dilated cardiomyopathy. AA aortic area, MA mitral area, PCG phonocardiogram, 1 first heart sound, 2 second heart sound
If second- or third-degree (complete) atrioventricular block with sinus rhythm is present, the patient will have intermittent “cannon” A waves in the jugular venous pulse (Fig. 15.24). Patients with atrial fibrillation do not have an A wave in the jugular venous pulse but, instead, have an irregularly irregular pulse. Those with important tricuspid insufficiency have prominent V waves in their jugular venous pulse (Fig. 15.25). Patients with right ventricular (RV) failure have an increased jugular venous pressure. They may also have hepatomegaly and right upper quadrant tenderness if acute hepatic congestion develops, ascites if the RV failure is severe, and peripheral or sacral edema.
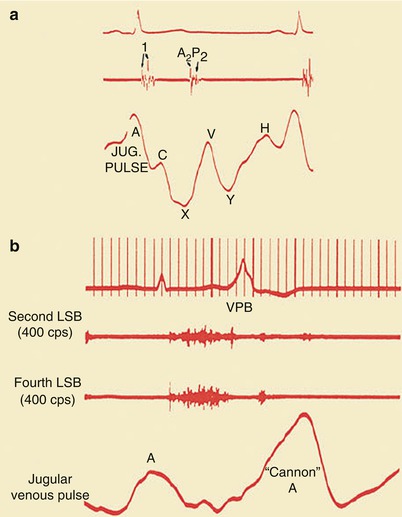
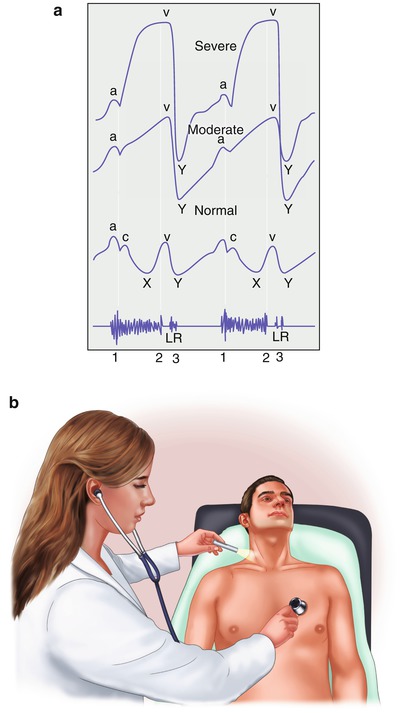
Fig. 15.24
(a) Normal jugular (jug.) venous pulse configuration. Note the prominence of the A wave and the fact that the X trough is deeper than the Y trough. Heart sounds and the ECG are shown (top). (b) A “cannon” A wave in the jugular venous pulse. Heart sounds are shown along with the ECG (top). LSB left sternal border, VPB ventricular premature beat
Fig. 15.25
(a) The normal jugular venous pulse (bottom) and the jugular venous pulse with moderate (middle) and severe tricuspid insufficiency (top). Note the marked prominence of the V wave with moderate and severe tricuspid insufficiency. The V wave correlates with the second heart sound (bottom, 2). The holosystolic murmur of tricuspid insufficiency is also shown (bottom). Typically, it becomes louder along the lower left sternal border with inspiration. L and R, respectively, denote timing of left and right ventricular third heart sounds (3). Third heart sounds occur with moderately severe and severe valvular regurgitation, and those emanating from the right side of the heart become louder with inspiration. (b) A preferred way to evaluate the jugular venous pulse waveform is to examine the right external jugular vein as shown and simultaneously listen to the heart sounds, timing the A wave with the first heart sound and the V wave with the second heart sound
With acute STEMI, a precordial “ectopic impulse” may sometimes be palpated over the left precordium, typically along the lower left sternal border or between the left sternal border and the apex. This impulse is caused by an increase in regional LV compliance within the area of injury, resulting in systolic distention or bulging (dyskinesia) of the injured heart segment. Over hours to a few days after an MI, the compliance of this region decreases (stiffness increases), and the systolic bulging is replaced by reduced (hypokinetic) or absent (akinetic) regional wall motion. If the systolic bulging persists, a ventricular aneurysm may be present.
Auscultation
Most patients with acute and chronic CAD (Fig. 15.26) will have an S4 that is usually soft and best heard by lightly applying the bell of the stethoscope over the middle and lower left region of the precordium. S4 is caused by a relatively forceful atrial contraction against a ventricle whose compliance is reduced as a consequence of increased ventricular stiffness caused by the physiologic effects of CAD.
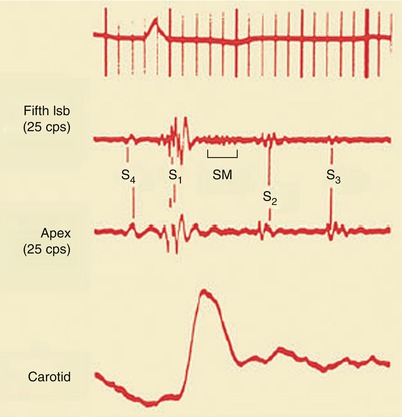
Fig. 15.26
Fourth heart sound (S 4 ), third heart sound (S 3 ), and a systolic ejection murmur (SM), representing mitral insufficiency. lsb left sternal border, S 1 first heart sound, S 2 second heart sound
When the mitral valve apparatus is damaged, a murmur of mitral insufficiency may be audible. These murmurs have variable auscultatory characteristics. They may be ejection murmurs in quality, peaking in intensity in mid- to late systole, or they may be holosystolic (Fig. 15.27). Intermittent myocardial ischemia with transient dysfunction of the posterior papillary muscle of the mitral valve leads to a murmur of papillary muscle dysfunction typically associated with mild to moderate mitral insufficiency. The murmur of papillary muscle dysfunction usually begins after the first heart sound (S1), peaks in mid- to late systole, and extends up to S2. It is heard at the cardiac apex and may radiate toward the left sternal border or into the axilla. If caused by intermittent myocardial ischemia, the murmur is transient. If caused by infarction of the papillary muscle, the murmur is usually permanent. Rupture of a papillary muscle with acute MI causes severe mitral insufficiency and often a soft apical holosystolic murmur. However, sometimes no murmur is audible with a ruptured papillary muscle, even though severe mitral insufficiency develops. Acute mitral insufficiency occurs most commonly in patients with inferior or lateral STEMIs and in those with NSTEMIs [151–157]. Similarly, those with inferior infarcts and structural damage to the tricuspid valve may develop tricuspid insufficiency.
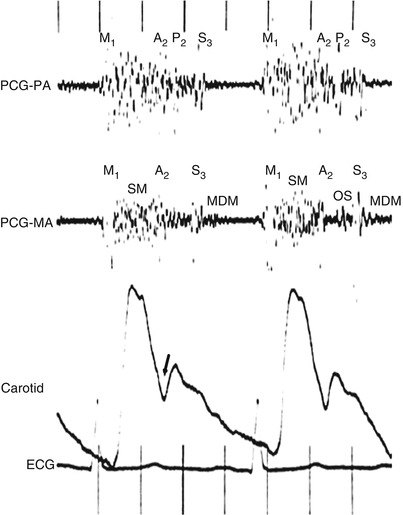
Fig. 15.27
Use of two simultaneous phonocardiograms (PCGs) to identify heart sounds in the patient with mitral regurgitation. A loud holosystolic murmur (SM) is noted at both the pulmonary artery (PCG-PA) and the mitral area (PCG-MA). The aortic component of the second heart sound (A 2 ) is masked by the latter part of the murmur, but it can be identified as the widely transmitted sound immediately preceding the incisura of the carotid pulse tracing (arrow). In this patient, mixed mitral regurgitation and stenosis coexist, and a mitral valve opening snap (OS) can be seen to occur slightly later than the second pulmonic heart sound (P 2 ) in the cardiac cycle. A prominent third sound (S 3 ) is seen in both PCG channels, and a mid-diastolic murmur (MDM) is present at the mitral area. M 1 first heart sound
Rupture of the interventricular septum occurs most commonly in patients with an acute anterior infarction, although it may also occur in patients with an inferior MI (Fig. 15.28) [158–167]. The murmur caused by a VSD is located along the lower left sternal border, is holosystolic, and is often associated with a left sternal border systolic thrill. As pulmonary artery pressure and vascular resistance increase, the systolic murmur of a VSD becomes shorter, ultimately disappearing altogether with the development of severe pulmonary hypertension (Fig. 15.28). Functionally large acute VSDs (with a pulmonary to systolic blood flow ratio of 1.5–1 or greater) should be closed, usually by means of surgical or interventional techniques, when CHF develops in a patient with MI; otherwise, rapidly progressive CHF and death may ensue. We recommend prompt coronary arteriography and repair of acute VSDs in patients who develop CHF.
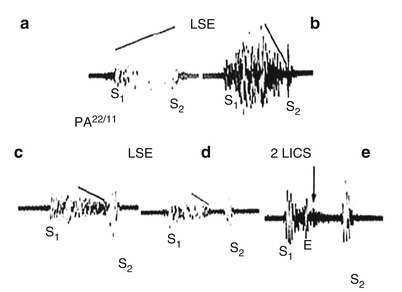
Fig. 15.28
The typical holosystolic murmur of a ventricular septal defect (VSD) and the influence of increasing pulmonary artery (PA) pressure on the murmur. (a) Murmur of a VSD with normal PA pressure. (b–e) The influence of increasing PA pressure on the murmur of a VSD shows that, with the development of severe pulmonary hypertension, the murmur becomes shorter and ejection-like in timing. With severe pulmonary hypertension, the systolic murmur of a VSD disappears. E ejection click, 2 LICS second left intercostal space, LSE lower sternal edge
A murmur of relative mitral or tricuspid insufficiency occurs in patients with LV or RV failure, respectively; such a murmur is the result of a spatial abnormality in the orientation of the papillary muscles of the mitral and tricuspid valves caused by marked dilatation of the left or right ventricle. With these entities, the mitral and tricuspid insufficiency is usually mild or moderate and diminishes in severity with diuresis and unloading therapy.
Third heart sounds occur in patients with acute MIs who have ventricular filling pressures (LV end-diastolic pressures) of 15 mmHg or greater and in those with moderately severe or severe mitral, aortic, or tricuspid valvular insufficiency (Fig. 15.26). S3 sounds are heard in patients with CHF and in those with rapid filling of the ventricles as a consequence of moderate or severe mitral, tricuspid, or aortic valve incompetence. These sounds may rarely be heard in young individuals (i.e., under 30 years of age) as a result of rapid filling of the ventricle.
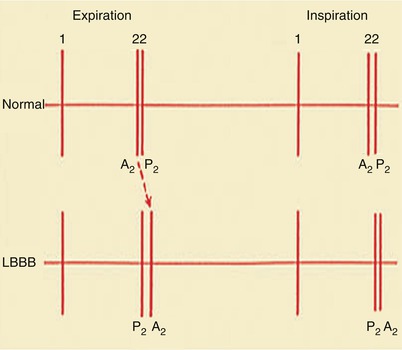
With inspiration, S2 sounds normally split into two components—the earlier aortic and the later pulmonary valve closure sound—because inspiration increases venous return to the right side of the heart and delays pulmonary valve closure (Fig. 15.29). However, the second sound may be paradoxically split (i.e., split wider during expiration) in patients experiencing angina pectoris, those with severe LV failure, systemic arterial hypertension, LBBB, or pacing from the right ventricle, and those with the various forms of LV outflow obstruction, including valvular, supravalvular, and subvalvular aortic stenosis (see Fig. 15.28). The pulmonary closure sound is increased in intensity in patients with moderately severe to severe pulmonary hypertension of any cause.