Fig. 1.1
Diagram of the usual anatomic distribution of the coronary arteries also showing a typical distribution of atherosclerotic plaques (dark areas). The blood supply to the heart is provided by the left and right coronary arteries and branches of these major vessels. The anterior wall of the left ventricle (LV) and the anterior portion of the interventricular septum are supplied by the left anterior descending coronary artery (LAD) and its diagonal and septal branches. The lateral wall of the LV is supplied by the left circumflex coronary artery (LCCA). The posterior wall of the left ventricle and posterior interventricular septum are usually perfused by the right coronary artery (RCA) which also supplies the right ventricle (From Willerson et al. [1]. Reprinted with permission from Wolters Kluwer)
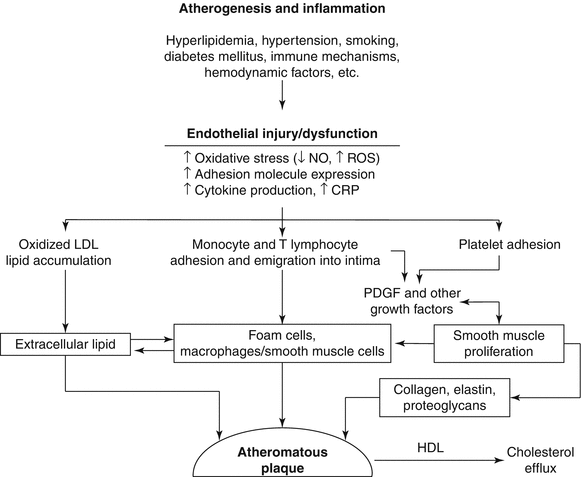
Fig. 1.2
Schematic diagram of postulated sequence of events and cellular interactions in atherogenesis. CRP C-reactive protein, NO nitric oxide, ROS reactive oxygen species, LDL low density lipoprotein, HDL high density lipoprotein (From Buja and McAllister [4]. Reprinted with permission from Springer)
Atherogenesis involves complex interactions between the vessel wall and soluble and formed elements of the blood (Fig. 1.2) [4–8]. Important factors in the initiation and growth of plaques are: (1) endothelial injury or dysfunction; (2) monocyte/macrophage accumulation; (3) influx of T lymphocytes; (4) platelet aggregation and attachment; (5) smooth muscle proliferation; (6) influx of plasma LDL; (7) oxidation of LDL; (8) progressive lipid accumulation in foam cells from uptake of oxidatively modified LDL; (9) apoptotic death of foam cells; (10) extracellular lipid deposition; and (11) hemodynamic influences related to blood pressure and pattern of blood flow.
Inflammation has a major role in the pathogenesis and clinical expression of atherosclerosis to the extent that atherosclerosis is now considered an inflammatory disease [4, 9–11]. Many key features of the inflammatory process have been found to occur in the development of atherosclerotic lesions (Fig. 1.3). Altered endothelial cells in lesion prone areas express a certain profile of adhesion molecules that lead to the recruitment, attachment and transmigration of blood monocytes and T lymphocytes. The local environment within the intima leads to transformation of the leukocytes into activated macrophages and T lymphocytes and promotes the chemical signaling between the two cell types. Cytokines produced by the macrophages create a proinflammatory environment that facilitates the recruitment of more inflammatory cells. The production of other mediators, including platelet derived growth factor (PDGF)-like molecules, leads to the recruitment and proliferation of vascular smooth muscle cells in the lesions. Production of superoxide anion and other reactive oxygen species leads to oxidation of low density lipoprotein (LDL). Subsequently, unregulated uptake of oxidized LDL by macrophages and smooth muscle cells via their scavenger receptors leads to formation of foam cells. As the plaques grow, a central core of necrotic debris and extracellular lipid develops related to apoptotic death of foam cells [8]. Smooth muscle cells lay down the connective tissue matrix comprising the fibrous capsule of the plaque. Macrophages produce collagenases and metalloprotenases that are important in the turnover of the connective tissue matrix [9, 11]. An excess of the degradative enzymes leads to degradation and thinning of the fibrous capsule. Plaques prone to rupture and ruptured plaques exhibit an inflammatory profile characterized by prominent numbers of macrophages and lymphocytes adjacent to the plaque capsule, increased expression of inflammatory mediators, increased local concentration of metalloproteinases, and prominent apoptosis of plaque cells [12, 13]. The inflammatory nature of atherosclerosis is manifest by the correlation of increased blood levels of inflammatory markers, especially high sensitivity C-reactive protein (hs CRP) and the subsequent development of atherosclerotic disease [14].
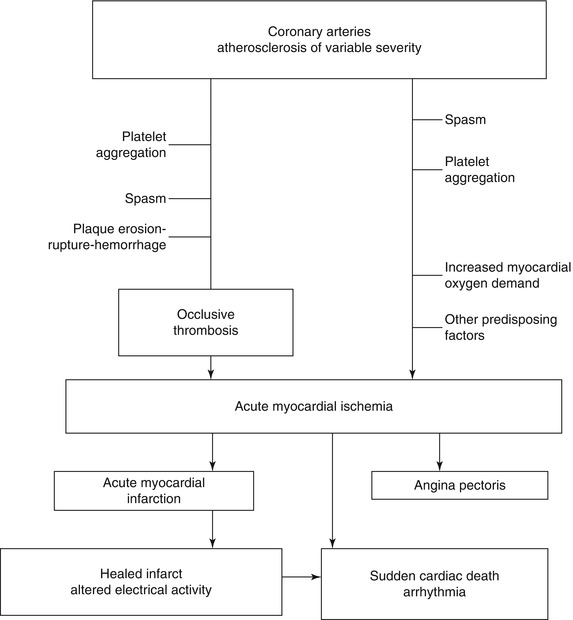
Fig. 1.3
Pathogenetic mechanisms of acute ischemic heart disease and potential clinical outcomes (From Buja and McAllister [3] (Reprinted with permission from Springer)
Initially, atherosclerosis is a focal disease. There is a predilection for formation of atherosclerotic plaques adjacent to branch points in areas of low-velocity flow and low shear stress adjacent to areas of high shear stress [6, 7, 15]. It is postulated that the flow patterns in such regions promote endothelial dysfunction and increased contact of endothelium with leukocytes and platelets. Established atherosclerosis involves all three layers of the arterial wall such that, in addition to intimal thickening, diseased areas exhibit medial degeneration and weakening and intimal fibrosis, with lymphocytic inflammatory infiltrates [3–5]. Areas of predilection for severe atherosclerosis in the coronary system include the proximal left anterior descending coronary artery and the proximal and distal right coronary arteries (Fig. 1.1).
Atherosclerotic disease leads to extensive remodeling of the vessel wall. Dilatation of the vessel occurs, in such a way that the lumen is maintained despite the presence of intimal plaque, which may develop in an eccentric or concentric pattern [13, 15–17]. Luminal narrowing occurs only when atherosclerotic disease is advanced. Approximately 50 % narrowing of luminal diameter (75 % luminal area) is needed before blood flow is affected. Areas of severe narrowing often develop in the setting of multifocal disease. All of these changes can lead to underestimation of the extent and severity of coronary atherosclerosis on visual inspection of coronary arteriograms (“luminograms”) [13]. Quantitative coronary arteriography can provide more objective measurements of absolute coronary dimensions and flow. Fractional flow reserve (FFR) provides the most accurate measure of the severity of a otenous [18].
Coronary Thrombosis and Other Acute Coronary Lesions
Acute ischemic heart disease is often initiated by acute changes superimposed on atherosclerotic plaques (Fig. 1.3) [1–3, 10–23]. The spectrum of thrombotic lesions includes platelet aggregates, mural (nonocclusive) thrombi, and occlusive thrombi (Figs. 1.4, 1.5, 1.6, and 1.7) [1–3, 10–23]. Major thrombi are frequently associated with significant disruptions of the plaque surface, which may appear as fissures, erosion, ulceration, or rupture (Figs. 1.4, 1.5, and 1.6). Coronary lesions that are particularly susceptible to such changes are atheromatous plaques with thin fibrous capsules and large cores of lipid-rich debris, and these lesions are designated as vulnerable plaques (Fig. 1.5) [12, 13, 22, 23]. However, endothelial erosion predisposing to thrombosis can involve fibrocellular plaques without significant lipid content [19, 20].

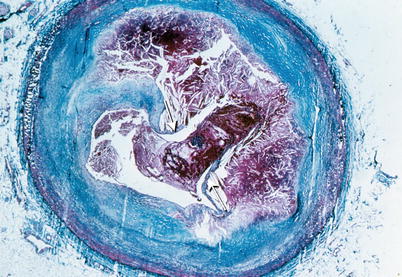
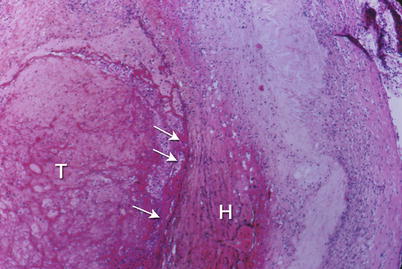
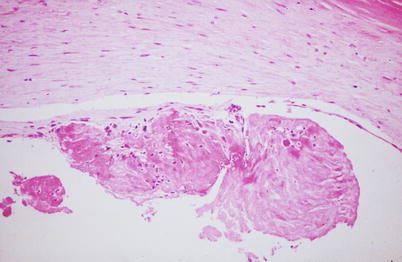
Fig. 1.4
Gross photograph of sections of a coronary artery with an atherosclerotic plaque and occlusive thrombus. A break in the plaque capsule has given rise to plaque hemorrhage (arrow) and occlusive thrombosis (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.5
Coronary artery shows a ruptured plaque capsule (arrows) and intraluminal and intraplaque thrombus. Low power photomicrograph (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.6
Coronary artery shows an erosion of the endothelial surface (arrows) leading to superficial hemorrhage in the plaque (H) and thrombosis (T) of the lumen. Low power photomicrograph (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.7
Coronary artery with a small mural thrombus attached to the surface of an atherosclerotic plaque. High power photomicrograph (From Buja and McAllister [3]. Reprinted with permission from Springer)
Inflammation adjacent to the plaque surface is important in the pathogenesis of alterations predisposing to thrombosis regardless of the plaque morphology [11–13]. Thus, high risk or vulnerable plaques are characterized by inflammation associated with a variety of plaque morphologies [19–21]. Factors that probably contribute to endothelial injury and disruption of the plaque surface include hemodynamic trauma, local attachment and activation of platelets and blood cells, inflammatory processes in the plaques, and cytotoxic effects of plaque contents, including metalloproteinases and other enzymes released from macrophages at sites of plaque rupture. The likely pathogenetic sequence of plaque rupture is endothelial injury, influx of blood components, increase in intraplaque pressure, and outward rupture of the fibrous capsule (Fig. 1.8) [1–3, 19–23]. Localized erosion and plaque fissuring also can give rise to platelet aggregation and thrombosis. Disruption of the plaque surface, by any mechanism, predisposes to formation of intraluminal and intramural (intraplaque) thrombus (Fig. 1.5).
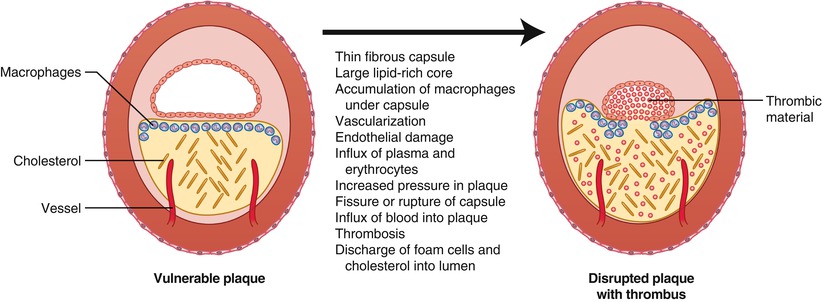
Fig. 1.8
Characteristics of the vulnerable plaque and mechanisms contributing to disruption of the plaque capsule and thrombosis (From Buja and McAllister [3]. Reprinted with permission from Springer)
Plaque hemorrhage may occur with or without thrombosis. Two mechanisms of intraplaque hemorrhage are influx of blood across the damaged endothelial surface of the plaque and influx of blood from small intraplaque vessels derived from the vasa vasorum. Intraplaque hemorrhage can increase plaque destabilization by contributing to the deposition of lipid, macrophage infiltration and enlargement of the necrotic core [1–3, 19–23]. Due to coronary remodeling, plaque rupture and occlusive thrombosis often occurs without prior significant luminal narrowing by the vulnerable plaque [22, 23].
Little information is available regarding anatomic correlates of coronary spasm [3, 24, 25]. Spasm is usually associated with atherosclerotic lesions but in some cases occurs without angiographically evident disease. Prominent adventitial inflammation and increased mast cells have been found to be more prevalent in coronary arteries of patients with a recent history of unstable angina pectoris at rest than in controls, further supporting a role for inflammatory mediators in the pathophysiology of coronary spasm [26]. Mechanisms for coronary artery spasm involve alterations of the vessel wall, including endothelial injury and hyperreactivity of vascular smooth muscle cells, coupled with a vasoconstrictive stimulus [25].
Nonatherosclerotic Coronary Vascular Diseases
In a small number of cases of ischemic heart disease, the coronary arteries are free of atherosclerosis and the clinical disease is related to some other condition. There is an interesting spectrum of nonatherosclerotic causes of ischemic heart disease, including congenital anomalies, dissection (Figs. 1.9 and 1.10), emboli, vasculitis, and other conditions of the coronary arteries [27]. Cocaine use can precipitate acute myocardial ischemia and infarction as a result of coronary spasm and/or thrombosis [28].
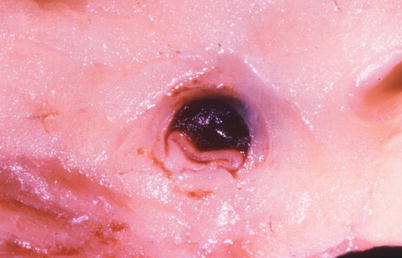
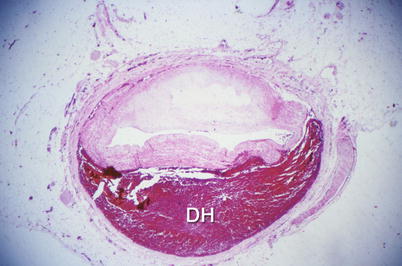
Fig. 1.9
Gross photograph of a spontaneously dissected coronary artery with a hematoma in the wall and marked compression of the lumen (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.10
Coronary artery with spontaneous dissecting hematoma (H) in the vessel wall. Low power photomicrograph (From Buja and McAllister [3]. Reprinted with permission from Springer)
Pathology of Angina Pectoris and Sudden Cardiac Death
The usual pathologic correlate of angina pectoris is coronary atherosclerosis with significant luminal narrowing of one or more of the major coronary arteries [1–3, 19–21]. However, there is considerable variation in the anatomic extent of large vessel CAD associated with the development of symptomatic ischemic heart disease. The variability is influenced by a number of interrelated factors, including the rate of progression of large vessel disease and the development of the coronary collateral circulation. Depending on the extent of coronary collateral blood flow, coronary occlusion may lead to a major myocardial infarct (MI) or to little or no myocardial damage. Thus, the myocardium can develop a spectrum of responses to alterations in coronary perfusion (Table 1.1) [29].
Table 1.1
Myocardial responses to alterations in coronary perfusion
Dysfunction during transient ischemia (angina pectoris) |
Stunning following acute ischemia |
Hibernation during chronic ischemia |
Ischemic preconditioning |
Against stunning |
Against infarction |
Acute myocardial infarction (AMI) |
Subendocardial, transmural |
Extension, expansion |
Complications |
Reperfusion effects |
On myocardial function |
On myocardial injury |
Postconditioning |
Remodeling |
Chronic ischemic heart disease |
Ischemic cardiomyopathy |
Unstable angina pectoris and related syndromes (preinfarction angina, coronary insufficiency) are associated with a high incidence of acute alterations of plaques (“unstable plaques”) with superimposed thrombotic lesions, usually platelet aggregates or nonocclusive thrombi, as well as platelet aggregates in the microcirculation of the myocardium [1–3, 19–21]. The accumulation of macrophages at sites of unstable, vulnerable plaques, indicates an inflammatory component to these vascular lesions, has also been demonstrated [11–13].
Coronary atherosclerosis is the most frequent anatomic substrate of sudden cardiac death [30, 31]. In large series, approximately 90 % of cases exhibit significant atherosclerotic narrowing of at least one coronary artery [31]. Many of the cases also show evidence of previous myocardial injury, manifest as multifocal myocardial scarring and/or healed infarction [31]. Although most cases do not exhibit an anatomically demonstrable acute MI, a subset of cases of sudden death are related to acute MI [31–34]. There is considerable variability in the reported incidence of acute plaque alterations and thrombotic lesions [31–34]. However, evidence of coronary plaque disruption and thrombosis has been documented in a significant subset of patients, particularly those with a prior history of unstable angina pectoris, an acute MI or single vessel disease [31–34]. Such patients also frequently show evidence of platelet aggregation in the coronary microcirculation.
Women and men exhibit differences regarding sudden cardiac death [3, 33, 34]. Sudden cardiac death occurs more frequently in men than women. Differences in coronary lesions also have been observed, with superficial plaque erosion rather than plaque rupture occurring more frequently in younger individuals and women. There is evidence of a higher incidence of plaque rupture in men dying suddenly during exertion than in men dying suddenly at rest. Furthermore, plaque rupture with exertion is characterized by a relatively thin fibrous capsule, relatively numerous vasa vasorum, and rupture in mid-cap, whereas plaque rupture at rest tends to occur at the shoulder region of the fibrous cap. In summary, clinicopathologic studies support the concept of three major mechanisms of sudden cardiac death: ischemia-induced ventricular arrhythmia without acute MI; acute MI with ventricular arrhythmia; and primary ventricular arrhythmia associated with old myocardial damage and altered electrical conduction (Fig. 1.3) [30, 31].
Pathology of Acute Myocardial Infarction
MI is defined as the death of heart muscle resulting from severe, prolonged ischemia [1–3, 35–40]. MIs usually involve the LV. The relatively unusual RV infarcts occur in association with LV infarcts, particularly posterior transmural LV infarcts, or as isolated entities, usually in association with pulmonary hypertension. Most MIs are confined to the distribution of a single coronary artery and are designated as anterior, anteroseptal, lateral, and posteroinferior. Multiregional infarcts also occur. MIs are designated as subendocardial (non-Q-wave) when the necrosis is limited to the inner half of the ventricular wall (Fig. 1.11) or transmural (Q-wave) when the necrosis involves not only the inner half but significant amounts of the outer half of the ventricular wall (Figs. 1.12 and 1.13). The electrocardiographic (ECG) correlates are the ST segment elevation with subsequent Q-wave pattern for transmural infarcts (STEMI) and the ST segment depression without Q-wave pattern for subendocardial infarcts (NSTEMI) [1–3, 35–40].
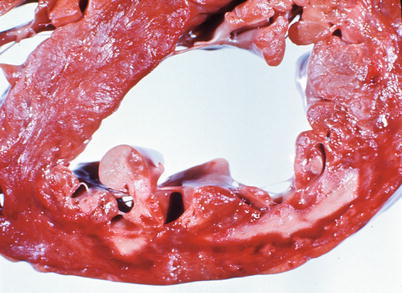
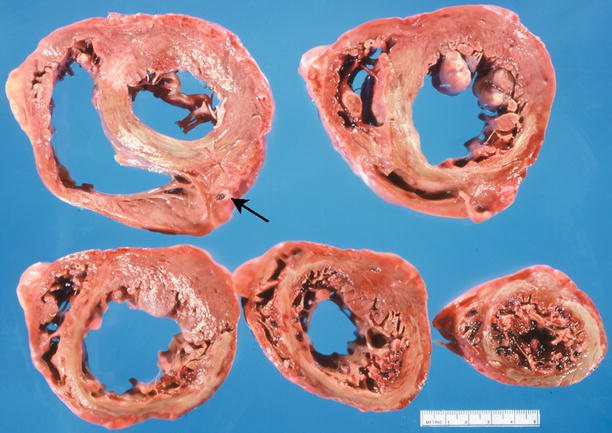
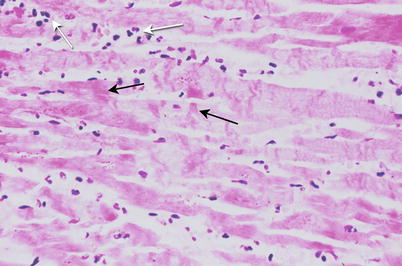
Fig. 1.11
Heart section demonstrates an acute subendocardial myocardial infarct involving the anterior left ventricle. The necrotic subendocardial myocardium is pale yellow and rimed by a red area of congestion (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.12
Transverse sections of the heart demonstrate a large acute transmural anteroseptal myocardial infarct (yellow area) related to an occlusive thrombus (arrow) of the proximal left anterior descending coronary artery (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.13
Acutely infarcted myocardium contains necrotic myocytes with contraction bands (black arrows) and an infiltrate of neutrophils (white arrows). High magnification micrograph (From Buja and McAllister [3]. Reprinted with permission from Springer)
The overall incidence of occlusive coronary thrombosis and associated plaque fissure or rupture is high (greater than 75 %) for acute MI [1–3]. The thrombus typically involves the major coronary artery in the distribution of the infarcted myocardium. However, there is a significant difference in incidence of thrombosis according to the type of infarct. In autopsy studies, occlusive coronary thrombi are found in more than 90 % of cases of transmural (STEMI) MI but in only about one-third of cases of subendocardial (NSTEMI) MI [1–3]. Subendocardial MI without occlusive thrombosis is related to the influence of other factors, such as more subtle coronary lesions (platelet aggregation, nonocclusive thrombi) and/or factors that increase myocardial oxygen demand (e.g., aortic stenosis, systemic hypertension, cardiac hypertrophy, excessive stress, or exertion) (Fig. 1.3). The occurrence of subendocardial MI without occlusive thrombosis highlights the increased susceptibility of the human subendocardium to ischemic injury. This susceptibility is caused by a more tenuous oxygen supply-demand balance in this region versus the subepicardium. This, in turn, is related to the pattern of distribution of the collateral circulation and to local metabolic differences in subendocardial versus subepicardial myocytes [1–3].
The major complications of acute MI are infarct expansion (shape change leading to stretching and thinning of the ventricular wall), infarct extension (additional necrosis), cardiogenic shock and/or recurrent ventricular arrhythmias related to large infarct size (generally greater than 33–40 % of LV mass), papillary muscle dysfunction, papillary muscle rupture (Fig. 1.14), external cardiac rupture (Fig. 1.15), ventricular aneurysm (Fig. 1.16), ventricular pseudoaneurysm (due to sealing off of a relatively slowly evolving rupture), ventricular septal rupture, pericarditis (nonspecific and autoimmune, e.g., Dressler’s syndrome) systemic embolization from an LV mural thrombus, and pulmonary thromboembolism [38–40].
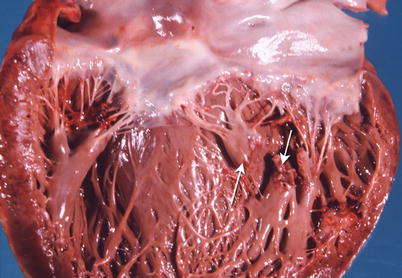
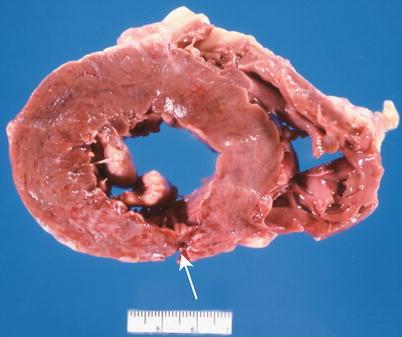
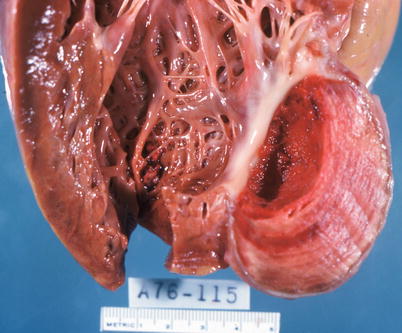
Fig. 1.14
Heart shows a rupture of the posterior papillary muscle (arrows) due to an acute myocardial infarct (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.15
Section of heart shows an acute transmural posterior myocardial infarct with an external rupture site (arrow) (From Buja and McAllister [3]. Reprinted with permission from Springer)
Fig. 1.16
Left ventricular aneurysm with mural thrombus resulting from healing of a transmural myocardial infarct (From Buja and McAllister [3]. Reprinted with permission from Springer)
The risk for infarct rupture is significant during the first week of MI before significant organization of the necrotic tissue [38–40]. Healing of MI involves neutrophil infiltration followed by formation of granulation tissue. Granulation tissue is grossly visible at approximately 10 days and completely replaces the necrotic tissue by 2–3 weeks. Thereafter, the granulation tissue is converted to a dense scar; this process is completed in 2–3 months.
Pathogenesis of Myocardial Ischemic Injury
The pathogenesis of ischemic myocardial cell injury and necrosis involves complex metabolic and structural alterations induced by severely reduced blood flow (Fig. 1.17) [29, 35–37, 42–44]. As a result of oxygen deprivation, mitochondrial oxidative phosphorylation rapidly ceases, with resultant loss of the major source of ATP synthesis. Initially, there is a compensatory increase in anaerobic glycolysis. However, this process leads to accumulation of hydrogen ions and lactate, with a resultant intracellular acidosis and inhibition of glycolysis as well as mitochondrial fatty acid and energy metabolism [29, 35].
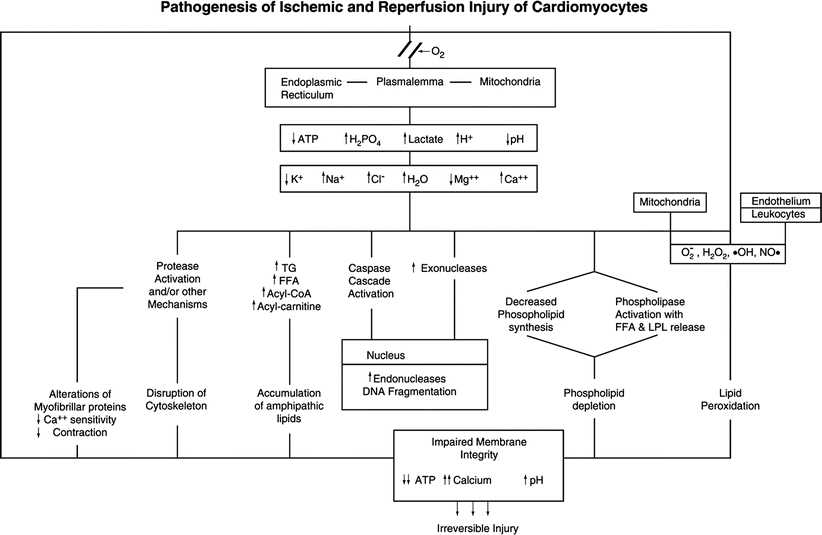
Fig. 1.17
Postulated sequence of alterations involved in the pathogenesis of irreversible myocardial ischemic injury and reperfusion injury. The potential exists for activation of apoptotic (caspase-mediated) and oncotic (multisystem metabolic) pathways in the same perturbed cardiomyocytes. Oxygen deficiency induces metabolic changes, including decreased ATP, decreased pH, and lactate accumulation, in ischemic myocytes. The irreversible phase of oncotic injury in energy depleted cardiomyocytes is mediated by severe membrane damage produced by phospholipid loss, lipid peroxidation and cytoskeletal damage. In cardiomyocytes with some preservation of ATP, activation of the caspase cascade can lead to a predominant apoptotic type of cell death. FFA free fatty acids, LPL lysophospolipids, TG triglycerides (From Buja and Weerashinghe [41]. Reprinted with permission from Elsevier Limited)
The metabolic alterations are associated with inhibition of contraction (excitation-contraction uncoupling) and associated alterations in ionic transport systems located in the sarcolemma and organellar membranes [29]. The initial alteration is loss of intracellular K+ due to increased efflux of the ion. Although the mechanism is unclear, it may involve activation of ATP-dependent K+ channels due to change in the ATP/ADP ratio or a mechanism to reduce osmotic load. Another early change is an increase in free Mg2+, followed by a decrease in total Mg2+. Once ATP decreases substantially, the Na+, K+ -ATPase is inhibited, resulting in a further loss of K+ and an increase in Na+. The accompanying influx of extracellular fluid leads to cell swelling. An early increase in cytosolic Ca2+ also occurs as the result of multifactorial changes in transport systems of the sarcolemma and sarcoplasmic reticulum [29]. Alterations in myofibrillar proteins leads to decreased sensitivity to Ca2+ resulting in impaired contractility in spite of the elevated cytosolic Ca2+ [44, 45]. As a result of the metabolic derangements, a key transitional event involves the opening of the mitochondrial permeability transition pore (mPTP) in the inner membrane of the mitochondria. Opening of the mPTP causes immediate loss of the electrical potential difference across the inner membrane (∆Ψm) leading to cessation of ATP synthesis, influx of solute, and severe mitochondrial swelling [45, 46].
Advanced ischemic myocardial cell injury is mediated by progressive membrane damage involving several contributory factors (Fig. 1.17) [29, 37]. Calcium accumulation or other metabolic changes lead to phospholipase activation and resultant phospholipid degradation and release of lysophospholipids and free fatty acids. Impaired mitochondrial fatty acid metabolism leads to accumulation of various lipid species, including long-chain acyl CoA and acyl carnitine which, together with products of phospholipid degradation, can incorporate into membranes and impair their function. Free radicals, including toxic oxygen species, are generated from ischemic myocytes, ischemic endothelium, and activated leukocytes. These toxic chemicals induce peroxidative damage to fatty acids of membrane phospholipids. Probably as a result of protease activation, cytoskeletal filaments, which normally anchor the sarcolemma to adjacent myofibrils, become damaged, and their anchoring and stabilizing effect on the sarcolemma is lost. All of these changes lead to a progressive increase in membrane permeability, further derangements in the intracellular ionic milieu, and ATP exhaustion. The terminal event in initiating irreversible myocyte injury is physical disruption of the sarcolemma of the swollen myocyte [29, 37]. The progression and pattern of injury are impacted importantly by the extent of reperfusion [41, 47–49].
The sequence of abnormalities described above constitute the well documented pathophysiological basis of cell injury leading to cell death in cardiac myocytes subjected to a major ischemic or hypoxic insult. However, recent discoveries have indicated that cardiomyocytes, as with other cells and tissues, are subject to three major modes of cell injury and death, namely, apoptosis (programmed cell death Type I), dysregulated autophagy (programmed cell death Type II), and oncosis (apparently passive cell death) [50–54]. Non-apoptotic cell death is commonly referred to as necrosis. But necrosis more precisely is defined as the final common pathway of cell degeneration following any mode of cell injury and death. Non-apoptotic oncotic cell death is increasingly being recognized to follow integrated and regulated steps, i.e., a different form of programmed injury. This has given rise to the use of new terms of programmed necrosis, necroptosis and Type III necrosis to characterize this process [52–54].
Following the recognition of apoptosis as a major and distinctive mode of cell death, reports have been published implicating apoptosis in myocardial infarction, reperfusion injury, and other forms of cardiovascular pathology [56]. Apoptosis is characterized by a series of integrated (programmed) molecular and biochemical events, including: (a) gene activation (programmed cell death); (b) perturbations of mitochondria, including membrane permeability transition and cytochrome c release; (c) activation of a cascade of cytosolic aspartate-specific cysteine proteases (caspases); (d) endonuclease activation leading to double-stranded DNA fragmentation; and (e) altered phospholipid distribution of cell membranes and other surface properties with preservation of selective membrane permeability [57–61]. Apoptosis can be triggered by activation of a death receptor pathway or a mitochondrial pathway [57–61]. Apoptosis is also characterized by distinctive morphological alterations featuring cell and nuclear shrinkage and fragmentation, with subsequent phagocytosis of apoptotic bodies by adjacent viable cells without exudative inflammation [45, 50, 51].
Apoptosis and oncosis are mediated by distinct but overlapping pathways involving cell surface death receptors, mitochondria and endoplasmic reticulum. The critical event in apoptosis is mitochondrial outer membrane permeabilization (MOMP) which promotes release of cytochrome c and other molecules leading to caspase activation. Conversely, the critical mitochondrial event in oncosis is early opening of the mitochondrial permeability transition pore (mPTP) in the inner membrane, which occurs without cytochrome c release [52–54]. The rate and magnitude of ATP reduction may be a critical determinant of whether an injured myocyte progresses to death by apoptosis or oncosis. Studies using caspase inhibitors and mutant mouse models indicate that apoptosis as well as oncosis contribute to the overall magnitude of ischemic necrosis, with the dominant mechanism being apoptosis and oncosis in approximately equal percentage of ischemic myocytes, respectively [41, 45, 60]. Apoptotic and oncotic mechanisms likely are operative in the same myocytes during progression to irreversible ischemic injury and necrosis [51]. The same injured myocyte may undergo activation of the apoptotic pathway with activation of the caspase cascade followed by activation of oncotic mechanisms leading to cell membrane damage and terminal cell swelling and rupture (Fig. 1.17) [41]. Thus, the cell injury and death induced by severe coronary blood flow reduction appears to represent a hybrid pattern best designated as ischemic cell injury and cell death. In contrast to primary oncotic necroptosis, secondary necrotic change follows apoptosis if the rapid removal of apoptotic bodies does not occur. In this event, necrotic events, such as loss of the mitochondrial membrane potential, can occur in close temporal relationship to cytochrome c release from the mitochondria [52–54].
Recently, autophagy has been identified as another mode of cell modulation that can contribute to various forms of ischemic injury [61, 62]. Autophagy, or macroautophagy, is an intracellular catabolic process involving the incorporation of cytoplasmic proteins and intracellular organelles in double membrane-lined autophagosomes that fuse with lysosomes followed by degradation of the material. Controlled autophagy is important in the normal turnover of organelles. However, unregulated autophagy can lead to a form of cell death. Interactions between autophagy and apoptosis contribute to the myocardial response to acute and chronic ischemic injury [61, 62].
Determinants of Infarct Development and Size and Remodeling
After coronary artery occlusion, the myocardium can withstand up to about 20 min of severe ischemia without developing irreversible injury. However, after about 20–30 min of severe ischemia, irreversible myocardial injury begins [36, 37]. The subsequent degenerative changes give rise to recognizable myocardial necrosis (Fig. 1.13). In the human and dog, myocardial necrosis first appears in the ischemic subendocardium, because this area usually has a more severe reduction in perfusion compared with the subepicardium. Over the ensuing 3–4 h, irreversible myocardial injury progresses in a wavefront pattern from the subendocardium into the subepicardium (Fig. 1.18) [36]. In the experimental animal and probably in humans as well, most MIs are completed within approximately 4 h after the onset of coronary occlusion. However, a slower pattern of evolution of MI can occur when the coronary collateral perfusion is abundant and/or when the stimulus for myocardial ischemia is intermittent (e.g., in the case of episodes of intermittent platelet aggregation before occlusive thrombosis).
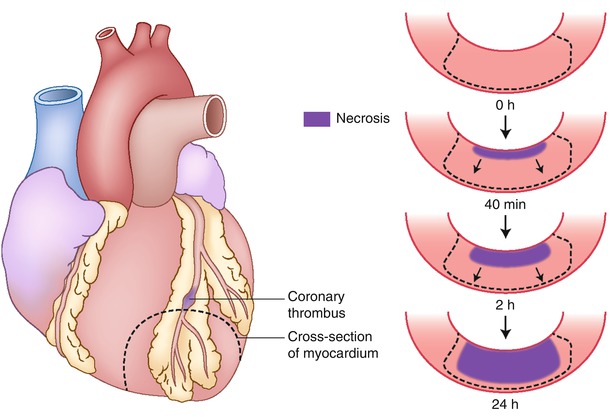
Fig. 1.18
Progression of cell death versus time as a wavefront of necrosis at various time intervals after coronary occlusion. Necrosis occurs first in the subendocardial myocardium. With longer intervals of occlusion, a wavefront of cell death moves from the subendocardial zone across the wall to involve progressively more of the transmural thickness of the ischemic zone. In contrast, the lateral margins in the subendocardial region of the infarct are established as early as 40 min after occlusion and are sharply defined by the anatomic boundaries of the ischemic bed (From Buja and McAllister [3]. Reprinted with permission from Springer)
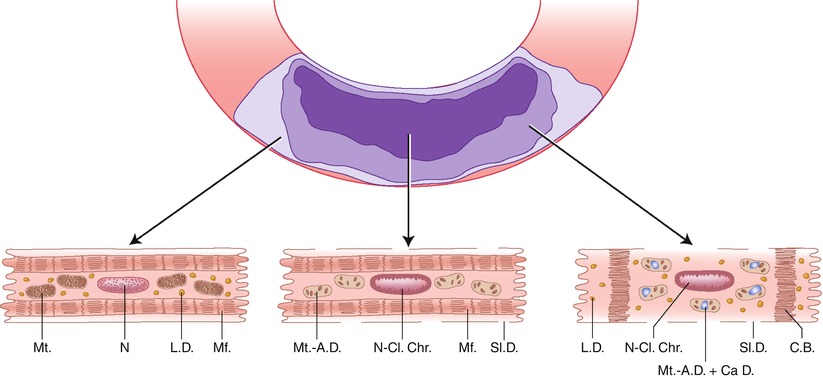
Established myocardial infarcts have distinct central and peripheral regions (Fig. 1.19) [29, 37]. In the central zone of severe ischemia, the necrotic myocytes exhibit clear sarcoplasm with separation of organelles (evidence of edema); clumped nuclear chromatin, stretched myofibrils with widened I-bands, swollen mitochondria containing amorphous matrix (flocculent) densities composed of denatured lipid and protein and linear densities representing fused cristae, and defects (holes) in the sarcolemma. In the peripheral region of an infarct, which has some degree of collateral perfusion, many necrotic myocytes exhibit edematous sarcoplasm; disruption of the myofibrils with the formation of dense transverse (contraction) bands, swollen mitochondria containing calcium phosphate deposits as well as amorphous matrix densities, variable amounts of lipid droplets, and clumped nuclear chromatin. A third population of cells at the outermost periphery of infarcts contains excess numbers of lipid droplets but does not exhibit the features of irreversible injury just described. The pattern of injury seen in the infarcted periphery is also characteristic of myocardial injury produced by temporary coronary occlusion followed by reperfusion. In general, the most reliable ultrastructural features of irreversible injury are the amorphous matrix densities in the mitochondria and the sarcolemmal defects. The ultrastructural features correlate with the three histopathological patterns of coagulation necrosis, coagulative myocytolysis (contraction band necrosis) and colliquative myocytolysis (myocytolysis) [1, 29].