Contemporary Surgical Therapy for Tetralogy of Fallot
Charles D. Fraser Jr.
Muhammad S. Khan
SECTION I
An Overview
The development of effective treatment strategies for variants of tetralogy of Fallot (TOF) is inextricably linked to modern advances in the surgical treatment of congenital heart disease. In many ways, this lesion should be viewed as the benchmark against which other treatment strategies for congenital cardiac defects are compared. In fact, no other condition offers the scope and breadth of treatment history, particularly if one considers those congenital abnormalities involving biventricular circulation and abnormalities of connection of the right ventricle to the pulmonary arteries. In this regard, it is incumbent upon the current era of cardiac surgeons to pay particularly close attention to the evolution of strategies for surgical treatment of TOF and, very specifically, contemporary outcomes as well as long-term follow-up series. Quite clearly, these data should be used to further refine treatment strategies in the present and future eras.
Prior to the advent of surgical therapy, the natural history of untreated TOF offered a dismal prognosis for patients. In the early part of the 20th century, therapeutic options were meager and included avoidance of stressful situations and, in rare circumstances, phlebotomy for those patients with advanced chronic polycythemia.
Without doubt, the field of palliative congenital heart surgery was ushered in with the advent of the successful application of a surgically created systemic to pulmonary artery shunt (Fig. 93.1). The Blalock shunt was enormously successful in augmenting pulmonary blood flow, bringing significant symptomatic, and in some cases durable relief to the chronic sequelae of cyanosis (Fig. 93.2). This was an enormous conceptual advance in surgery and arguably should be heralded as transformative in its impact on surgical thinking about developing other strategies to treat congenital heart disease. The much heralded and lingering debate over credit for the development of this operation probably should be put to rest, in which it appears that the historical record is quite clear. During his tenure on the faculty at Vanderbilt University School of Medicine in Nashville, TN, Dr. Alfred Blalock developed a renowned surgical laboratory, primarily involved in the investigation of shock. In that context, Blalock was introduced to a young man who would become part of one of the most productive surgical investigational relationships of that century when he hired Mr. Vivien Thomas to work in his lab. Much has been written about this storied relationship and the reader is referred to the bibliography for further references of the details of the relationship. However, it is important to emphasize that Mr. Thomas was a disadvantaged African-American man, denied access to formal medical education because of economic hardships during the post-Depression era in the southern United States. Through this providential relationship and the recognition of mutual strengths and aspirations, Blalock and Thomas effectively collaborated on a number of landmark investigations. Of special note is work that was published by Blalock in 1933 on the prospect of the creation of a model of pulmonary arterial hypertension in the canine model. The surgical technique involved the division of the left subclavian artery and a direct end-to-end anastomosis into the divided left pulmonary artery. While unsuccessful in creating hypertension in this animal model because of the extremely low pulmonary vascular resistance in the canine, without doubt the technical nuances of the delicate systemic to pulmonary artery anastomosis were worked out in the research laboratory at Vanderbilt University. After being recruited to become the Chairman of Surgery at the Johns Hopkins Hospital, Dr. Blalock collaborated with Dr. Helen Taussig in applying the previously developed surgical technique to the palliation of children with disorders limiting pulmonary blood flow. The much heralded first clinical application and the series thereafter established the Johns Hopkins Hospital as the early epicenter of surgical treatment of congenital heart disease. The shared credit for this successful series of operations rests with Blalock, Taussig, as well as Thomas and the surgical and medical practitioners of the current era should respectfully acknowledge the contributions of all three of these remarkable individuals and their impact on the treatment of children with heart disease.
Emboldened by the early successes with surgical palliation, Drs. John W. Kirklin at the Mayo clinic and C. Walton Lillehei at the University of Minnesota began to develop successful operations to directly correct the intracardiac pathology in TOF. The first series of surgically corrected patients were reported by these two pioneers in the mid-to-late 1950s. While surgical techniques have been improved and perioperative management has afforded patients a very favorable progress in the current era, it must be emphasized that the cohort of these earliest successful surgeries, now being followed for almost 60 years, represents an unusual, perhaps unique, window of observation into the natural history of surgically repaired congenital heart disease. In the context of TOF, this observation cannot be overemphasized and will be the focus of subsequent commentary in this chapter as it relates to timing and approach for surgery.
Anatomy and Physiology
By way of definition, this chapter will be limited to discussion of classic TOF. This lesion, originally described by Étienne-Louis Arthur Fallot in a postmortem series in late 1888, represents the prototypical cyanotic congenital heart disease. As is well articulated by leading cardiac morphologists, including Drs. Stella and Richard Van Praagh at Boston Children’s Hospital and Dr. Robert H. Anderson at the Royal Brompton Hospital and the
National Heart Trust of the United Kingdom, the central morphologic feature of TOF relates to anterior malalignment of the infundibular septum relative to the conotruncus resulting in a large subaortic ventricular septal defect (“malalignment VSD”) and varying degrees of right ventricular outflow tract obstruction (Fig. 93.3). The additional classic elements of TOF include secondary right ventricular hypertrophy and some degree of aortic override or malalignment relative to the interventricular septum. Again, for the purposes of this chapter, comments will be limited to conditions involving balanced ventricles with atrioventricular concordance, malalignment VSD, typically of perimembranous location and pressure nonrestrictive designation, and the presence of prograde pulmonary blood flow into identifiable native branch pulmonary arteries and the absence of major aortopulmonary collateral arteries and significant arborization abnormalities of the lungs which should be discussed in a separate chapter. Mention will be made, however, of patients with TOF with complete atrioventricular septal defect (AVSD) and double outlet right ventricle with subaortic VSD and RVOT obstruction (tetralogy-type double-outlet right ventricle). It is important to note that the degree of aortic override in this chapter will not be used to distinguish between TOF and double-outlet right ventricle, and all patients with the presence of aortomitral fibrous continuity irrespective of the degree of aortic override are to be considered in the spectrum of TOF (Fig. 93.4A, and 93.4B).
National Heart Trust of the United Kingdom, the central morphologic feature of TOF relates to anterior malalignment of the infundibular septum relative to the conotruncus resulting in a large subaortic ventricular septal defect (“malalignment VSD”) and varying degrees of right ventricular outflow tract obstruction (Fig. 93.3). The additional classic elements of TOF include secondary right ventricular hypertrophy and some degree of aortic override or malalignment relative to the interventricular septum. Again, for the purposes of this chapter, comments will be limited to conditions involving balanced ventricles with atrioventricular concordance, malalignment VSD, typically of perimembranous location and pressure nonrestrictive designation, and the presence of prograde pulmonary blood flow into identifiable native branch pulmonary arteries and the absence of major aortopulmonary collateral arteries and significant arborization abnormalities of the lungs which should be discussed in a separate chapter. Mention will be made, however, of patients with TOF with complete atrioventricular septal defect (AVSD) and double outlet right ventricle with subaortic VSD and RVOT obstruction (tetralogy-type double-outlet right ventricle). It is important to note that the degree of aortic override in this chapter will not be used to distinguish between TOF and double-outlet right ventricle, and all patients with the presence of aortomitral fibrous continuity irrespective of the degree of aortic override are to be considered in the spectrum of TOF (Fig. 93.4A, and 93.4B).
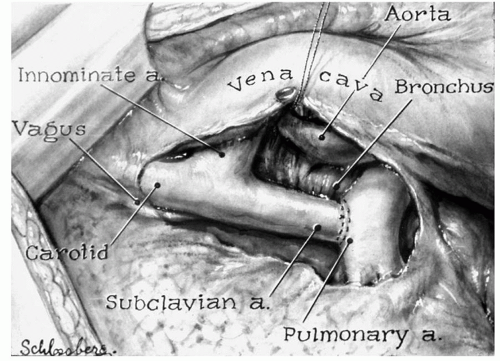 Fig. 93.1. The original illustration of the first BT shunt by Leon Schlossberg. (Image used with permission from The American College of Surgeons.) |
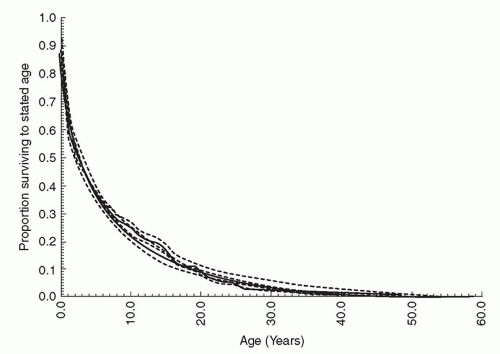 Fig. 93.2. The natural survival for unrepaired tetralogy of Fallot patients. (Reprinted with permission from Bertranou EG, Blackstone EH, Hazelrig JB, et al. Life expectancy without surgery in tetralogy of fallot. Am J Cardiol 1978;42(3):458-466.) |
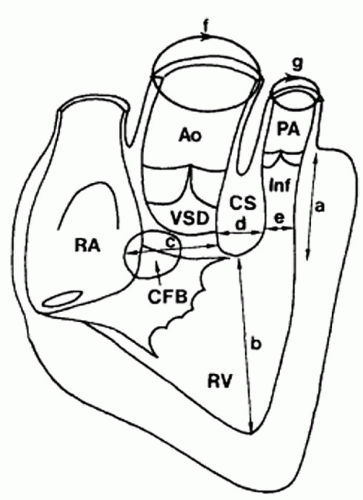 Fig. 93.3. A diagram showing the morphometric measurements of the tetralogy of Fallot heart. RA, right atrium; CFB, central fibrous body; RV, right ventricle; VSD, ventricular septal defect; Ao, aorta; CS, coronal septum; PA, pulmonary artery; Inf, infundibulum. The letters a through g indicate the dimensions recorded as described in the original article. (Diagrams used with permission from Becker AE, et al. Tetralogy of Fallot: a morphometric and geometry study. Am Card 1975;35:402-412.) |
Associated Conditions
Conditions frequently found associated with TOF include a persistent left superior vena cava, most commonly to an intact coronary sinus. In some patients, in addition to the outlet VSD there may be additional muscular VSDs. A patent foramen ovale is typically present. The ductal configuration is typically associated with normal ductal insertion, although it is not infrequent to observe ductal entrapment of the proximal left pulmonary artery which may ultimately lead to severe proximal left pulmonary artery stenosis or ductal coarctation.
A significant anomaly that may definitely affect the surgical approach is found in the presence of anomalous origin of the coronary arteries. The most frequent anomaly includes anomalous origin of the anterior descending coronary artery from the right coronary artery coursing across the subpulmonary infundibulum. Less frequently, a dominant left main coronary ostium gives rise to a large right coronary artery that originates from the left main trunk which travels around the pulmonary infundibulum and across the anterior surface of the RVOT. An associated right aortic arch is found in 25% of patients.
A significant anomaly that may definitely affect the surgical approach is found in the presence of anomalous origin of the coronary arteries. The most frequent anomaly includes anomalous origin of the anterior descending coronary artery from the right coronary artery coursing across the subpulmonary infundibulum. Less frequently, a dominant left main coronary ostium gives rise to a large right coronary artery that originates from the left main trunk which travels around the pulmonary infundibulum and across the anterior surface of the RVOT. An associated right aortic arch is found in 25% of patients.
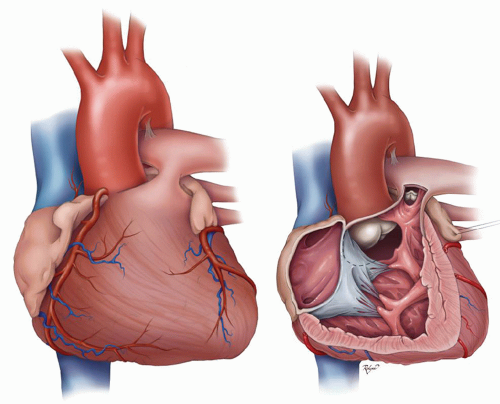 Fig. 93.4. The external (left) and internal (right) anatomy of the heart of a patient with tetralogy of Fallot. (Used with permission from Charles D. Fraser Jr., MD, Congenital Heart Surgery, Texas Children’s Hospital, Houston, TX) |
Embryology
For understanding the embryologic development of TOF, we refer the reader to the normal development of the heart. The truncus arteriosus is divided by the aorticopulmonary “spiral” septum that grows down from the midline at 7 to 8 weeks of fetal life to fuse with the ventricular septum. This essentially creates the two great vessels and also isolates the right and left ventricles. In the case of TOF, it is thought that the septum is shifted toward the right leading to three of the four tetralogy defects, that is overriding aorta, pulmonary stenosis, and a malalignment VSD. The fourth defect, that is right ventricular hypertrophy, develops to overcome resistance to blood flow caused by the combination of the earlier mentioned three defects as well as right ventricular pressure load.
TOF is seen in association with a number of chromosomal abnormalities and syndromes. Approximately 15% of patients with TOF have chromosome 22q11.2 deletion syndrome (the presentation of which includes DiGeorge syndrome), whereas 7% of Down’s syndrome (trisomy 21) patients have TOF. Other syndromic presentations include Pentalogy of Cantrell also known as thoraco abdominal syndrome, Alagille syndrome, and CHARGE (Coloboma of the eye/central nervous system anomalies, Heart defects, Atresia of choanae, Retardation of growth/development, Genital/urinary defects, Ear anomalies) syndrome. Certain genetic associations have also been found to exist in tetralogy patients that include mutation in NKX2.5, JAGGED1, ZFPM2/FOG, VEGF, and CHD7 genes.
Pathophysiology
The symptomatology of TOF presents a clinical spectrum related to the severity of RVOT obstruction at the infundibular, valvar, supravalvar, or branch pulmonary artery level. Newborns with severe infundibular and valvar obstruction may present with profound cyanosis and ductal-dependent pulmonary blood flow. However, the majority of patients remain relatively asymptomatic in the early period of life. Progressive hypertrophy of the right ventricle and infundibulum participate in a gradual progression toward a degree of right-to-left shunting at the level of the VSD and propensity for desaturation. Frank “tetralogy spells” representing an elevated catecholamine state and reactive infundibular obstruction may be severe but typically are not present until later in infancy. Nonetheless, the natural history in most patients portends a progressive propensity for cyanosis and ultimately, secondary morbid sequelae from that cause. On the other end of the spectrum, however, are the individuals who have anatomy morphologically consistent with TOF with anterior malalignment VSD, but in whom there is little to no obstruction of the pathway of pulmonary blood flow. These patients may present with normal saturations, and in some cases pulmonary overcirculation. These patients with “pink tetralogy” may actually present with signs and symptoms of congestive heart failure relative to a large left-to-right shunt (Fig. 93.5).
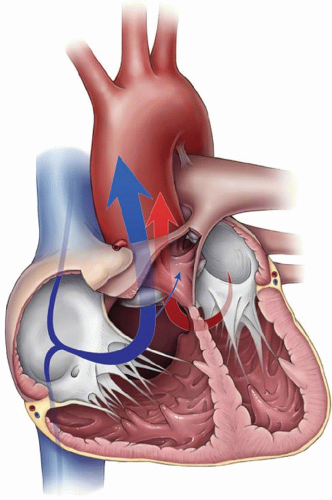 Fig. 93.5. A diagram documenting the directionality of pulmonary blood flow (blue arrows) during a tetralogy spell (red arrow indicates aortic blood flow). (Used with permission from Charles D. Fraser Jr., MD, Congenital Heart Surgery, Texas Children’s Hospital, Houston, TX.) |
SECTION II
Diagnostic Evaluation
Physical Examination
The level and degree of obstruction in the RVOT governs the symptomatology as well as the findings on physical examination. Those patients presenting with severe obstruction may have evidence of cyanosis, although this may, at times, be very difficult to discern, particularly in dark-skinned individuals. Additionally, many patients with cyanosis present with a dusky or even pale appearance, and the deep plethora of cyanosis is atypical, particularly in the current era with early intervention. Furthermore, the late sequelae of chronic cyanosis, including finger clubbing and profound nailbed cyanosis are also very uncommon in developed countries with the capability of early diagnosis and treatment of the patients. A point of emphasis merits attention here. In our observation, it is often very difficult for parents and even pediatricians to discern cyanosis on physical exam. Thus, in a conservative management strategy, it is unwise to rely too much on parents’ ability to detect desaturation.
Auscultation of patients with severe infundibular obstruction in patients with well-developed branch pulmonary arteries is often consistent with high-velocity flow through those structures, including highpitched systolic murmur and most often best auscultated at the left sternal border. Paradoxically, patients who present with profound hypercyanosis or frank spells often have diminution or even absence of the murmur as the pulmonary blood flow becomes severely restricted. On the other hand, patients with limited infundibular obstruction will often have very faint murmurs and may, in fact, present with signs of pulmonary overcirculation, including pulmonary crackles and a gallop rhythm.
Chest radiography classically reveals the coeur en sabot or boot-shaped heart configuration consistent with a limited pulmonary trunk shadow and a hypertrophied right ventricle and upturned apex. The arch sidedness may be right or left on chest radiography. In case of decreased pulmonary blood flow, the lung fields will show decreased vascular markings (oligemia).
In the current era, the primary diagnostic modality for patients presenting with TOF is transthoracic echocardiography. Important diagnostic features include the presence of prograde pulmonary artery flow, branch pulmonary artery size and distribution, precise delineation of VSD, and delineation of systemic and pulmonary venous connection. In those centers performing primary neonatal tetralogy repair for all patients, irrespective of symptomatology, the presence of an anomalous anterior descending coronary artery may be of significance and at some centers an extensive effort is made to discern this association, although at our hospital this has not been the standard approach. This issue will be expanded upon further later in this chapter.
The majority of patients with TOF do not require diagnostic cardiac catheterization. At our institution, catheterization now has limited application in TOF except in those cases where delineation of the pulmonary artery architecture has not been achieved through other imaging modalities, and it is perceived that this will have some relevance to the surgical timing and approach (see below). The theoretical application of interventional methods in the palliation of symptomatic TOF will be discussed in subsequent sections.
Computed Tomography Scan/Magnetic Resonance Imaging
These imaging modalities are of limited necessity in the primary diagnosis of TOF, although in some circumstances they may be used to ascertain the pulmonary artery architecture and origins and to screen for the presence of significant aortopulmonary collateral arteries.
Timing and Preoperative Care
The subjects of optimal timing and specific surgical technique remain sources of significant dialog and controversy in patients with symptomatic and asymptomatic TOF. It is this author’s opinion that an individualized strategy designed to optimize both early cardiac and neurodevelopmental function as well as long-term preservation of cardiac function and anatomy should be central features in any center’s perioperative management strategy. It is widely known that the late outcomes of patients with previously repaired TOF are predicated on important factors including tricuspid valve function, rhythm disturbance, right ventricular function, endocardial scarring, pulmonary artery distortion, pulmonary valve insufficiency, and chronic right ventricular pressurevolume overload as well as left ventricular dysfunction. Also of note, there is increasing evidence of late propensity of chronic aortic root dilation, progressive aortic valve insufficiency, and frank aneurysmal degeneration of the ascending aorta. As such, it appears logical that all current treatment strategies should take into account the prospect of affecting these late pathologic sequelae, and every effort should be expended to mitigate these circumstances. This is particularly true of right ventricular function where the timing and mode of therapy as well as the associated surgical “side-effects” must be taken into consideration.
In the current era of pediatric cardiac surgery, the substantial improvements in perioperative mortality rates has allowed for a refined focus on long-term neurodevelopmental outcome, and considerable ongoing effort is being expended in assessing the potential to optimize childhood neurologic potential. In this context, numerous investigators including our own institution have focused efforts on the risk profile for children born with congenital heart disease and whether the timing and mode of cardiac surgery can positively or negatively impact neurodevelopmental potential. Beyond the scope of this chapter, it is important to briefly note that current data would suggest that gestational age, perinatal stress, perinatal cyanosis, and application of cardiopulmonary bypass (including the adjunct of hypothermic circulatory arrest) have potential deleterious consequences on long-term neurodevelopmental outcomes. It is unclear at present whether further strategic refinement of timing and mode of repair can ultimately influence long-term neurodevelopmental potential in these patients, although it is highly intuitive that this is the case. In fact, the early arguments for promoting patients for aggressive primary neonatal repair of TOF were predicated on concerns about early perinatal exposure to cyanosis and the potential deleterious effects of that prospect. In contrast, however, recent evidence has also suggested that prudence is warranted in unnecessarily exposing newborns to the potential negative impact of perinatal stress. This is most markedly evident in the premature brain where numerous studies have demonstrated the vulnerability of the premature oligodendrocytes to stress. While definitive data are lacking and clearly enormous work needs to be done in carefully assessing the variables potentially relevant in brain injury and development, it is reasonable to conclude that surgeons in the current era must carefully consider the risk/benefit ratio of surgical approach, not only in terms of acute mortality but also the effects of neurodevelopmental potential.
Preoperative Preparation
Ductal-dependent newborns with severe RVOT obstruction and near pulmonary atresia being put forward for surgery will be maintained on a prostaglandin (PGE1) infusion until the time of operation. Typically, we favor a small bore percutaneous central catheter to provide durable intravenous access prior to surgery and the PGE1 is then discontinued at the initiation of cardiopulmonary bypass.
Infants and older children who have demonstrated a propensity toward cyanotic spells are often started on oral beta blockers by the referring cardiologist. In general, we have believed that patients who have been deemed to require beta-blocker therapy should be put forward for operation as soon as possible as hypercyanotic spells are unpredictable in many patients.
For patients in whom beta-blocker therapy has been initiated, there is a propensity toward perioperative sinus tachycardia, and our approach has been to continue the oral beta-blocker therapy up to the morning of surgery. These patients are also typically started on an infusion of short-acting beta blockers (Esmolol) prior to weaning from cardiopulmonary bypass. Frequently, during the preparatory stages there is a tendency for these patients to develop some degree of hyperthermia. This is, in general, not a favorable situation, and a mild degree of ambient hypothermia may be helpful in reducing the propensity for tachycardia and a catecholamine surge. In this setting, the anesthesiologist should work very closely with the surgeon to derive a management plan that is designed to limit the potential for hypercyanosis prior to the surgical correction. This may include oral or intravenous hydration, and as mentioned previously, intravenous beta-blocker therapy. Shunt-dependent newborns or infants referred for surgery should be admitted to hospital the night before operation and intravenous access should provide an opportunity for ongoing hydration as hypovolemia prior to operation may induce a cascade of problems, including acute shunt occlusion or hypercyanosis.
Newborn vs. Non-Neonatal Infant Repair
Much has been written about the appropriate timing for referring patients for surgery with TOF. In fact, it is believed in many institutions that the standard of care is a strict policy of referral for all patients with this diagnosis for primary newborn repair, irrespective of symptomatology and anatomy. While several notable investigators and institutions have opined that this approach now represents standard of care, this in fact is a highly debatable conclusion. A recent survey of data from the Society of Thoracic Surgeons confirms that the majority of TOF repairs are not performed in the newborn period. Nonetheless, there is much logic in considering primary repair in a newborn patient. Theoretically, this offers the potential to avoid or mitigate the propensity for cyanosis and the deleterious effects of that issue on organ system function, including neurologic development. As noted previously, this issue is not resolved. A compelling argument for promoting patients to surgery at the time of primary diagnosis rests in the implied follow-up responsibility should one not refer a patient at that time. Quite clearly, patients with ductal -dependent circulation require operation, and in later sections of this chapter, we will discuss that issue specifically. Nonetheless, most patients do not present with ductal dependent circulation and may be managed expectantly from a symptomatology viewpoint. It must be emphasized, however, that the management team, which should include a close interaction between the attending cardiologist and cardiac surgeon, must be highly vigilant for any evidence of progress toward cyanosis or spelling. This should also include careful education of the parents for diligent follow-up and timely intervention. As such, in those settings where these elements cannot be assured, and there is some risk of improper follow-up and timely referral for operation, it is entirely logical to promote these patients for surgery as a newborn. Whether or not this represents the best timing for surgery with regard to the long-term function of the right heart remains an issue of debate and should be the focus of ongoing longitudinal patient follow-up and refinement of surgical practice.
Our institutional policy at Texas Children’s Hospital since 1995 has been to perform non-neonatal infant repair of TOF except in those newborns who have ductal-dependent circulation or have morphology and symptomatology highly indicative of a propensity toward cyanotic spelling. In the current era, there appears to be no justification in delaying complete repair of TOF beyond infancy, and we typically favor performing this somewhere around 6 months of life (Fig. 93.6A-93.6D).
Late Presentation
While increasingly rare, there are patients who present undiagnosed late in childhood or even in adult life. These patients can, in fact, present significant management challenge. In particular, adults of advanced age with untreated TOF typically have developed extreme right ventricular hypertrophy, making intraoperative access challenging and perioperative management more complex.
 POSTOPERATIVE MANAGEMENT
POSTOPERATIVE MANAGEMENTSuccessful postoperative management of patients undergoing surgical repair of TOF is predicated on a thorough understanding of the nuances of acute postoperative right heart dysfunction. Successful complete repair of TOF involves elements of manipulation of right heart structures, which will be reflected on in more detail at a later point but that may, to a greater or lesser degree, exacerbate propensity for right heart dysfunction. In this regard, any management strategy that is additive to that in a negative sense will adversely impact patient prognosis through diminution of cardiac output and systemic venous congestion. In this regard, many surgeons, particularly those who strictly perform neonatal repair, will often leave a small patent foramen ovale or atrial septal defect to mitigate the effects of right heart distention and dysfunction. In our experience with selective, individualized management, this is not necessary, and we have not favored leaving an atrial level communication.
The management team in the intensive care unit should be vigilant about issues which affect right heart function and the management approach should be adjusted accordingly. In general, we favor an indwelling left atrial catheter as right and left heart filling pressures are often incongruous in the early perioperative period and accurate and effective fluid resuscitation must be evaluated in terms of right and left heart filling. There is a consistent tendency in practitioners less familiar with TOF to excessively volume resuscitate these patients which will exacerbate the propensity for right heart failure, hepatic congestion, pleural effusions, and prolonged recovery.
Inotropic management should be directed toward limiting tachycardia and systemic vasoconstriction. A vicious cycle that can be initiated by an uninformed or inexperienced management team is highly repeatable in patients with TOF. This, of course, is all the more prevalent in patients who have residual lesions, severe right heart dysfunction, tricuspid regurgitation, and dysrhythmia. Such patients may develop a central hyperthermia with
profound peripheral vasoconstriction. These patients often have a progressive tachycardia and the propensity for junctional ectopic tachycardia (JET) is quite significantly increased (to be discussed further at a later point). In this setting, the tendency is to escalate inotropic support and excessively volume resuscitate the patient. Our view is that every effort should be made to improve peripheral cardiac output and reduce the core hyperthermia. This may be effectively achieved with the addition of intravenous milrinone in addition to other systemic vasodilators. In this setting, we have used low-volume, room temperature peritoneal dialysis as well, usually with a reduction in the heart rate and improved diastolic filling, the cardiac output will improve. The goal is, of course, to avoid this cycle.
profound peripheral vasoconstriction. These patients often have a progressive tachycardia and the propensity for junctional ectopic tachycardia (JET) is quite significantly increased (to be discussed further at a later point). In this setting, the tendency is to escalate inotropic support and excessively volume resuscitate the patient. Our view is that every effort should be made to improve peripheral cardiac output and reduce the core hyperthermia. This may be effectively achieved with the addition of intravenous milrinone in addition to other systemic vasodilators. In this setting, we have used low-volume, room temperature peritoneal dialysis as well, usually with a reduction in the heart rate and improved diastolic filling, the cardiac output will improve. The goal is, of course, to avoid this cycle.
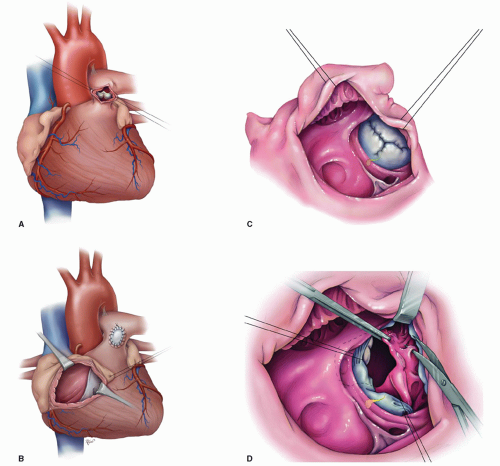 Fig. 93.6. (A-D
Get Clinical Tree app for offline access
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|