Considerations in the Design, Conduct, and Interpretation of Quantitative Clinical Evidence
Robert M. Califf
Eric J. Topol
Introduction
Medical practice has now enteredthe era of “evidence-based medicine,” characterized by an increasing societal belief that recommendations about the use of treatments in clinical practice should be based on scientific information rather than intuition or opinion. As our society’s tolerance for increasing the proportion of financial resources spent on medical care has been exhausted, the only rational way to allocate resources is to understand whether competing therapeutic approaches provide clinical benefit and, if so, the cost required to achieve that benefit. Simultaneous with the realization that expansion of medical financial resources is not limitless, the huge societal investment in biotechnology is beginning to pay off in the form of many potential new approaches to treating disease. Therefore, with current methodology, the need for evidence is increasing faster than the resources are being made available to perform the studies.
This accelerating pace of quantitative research creates a need for practicing physicians to understand the methods of evaluating clinical evidence well beyond the level taught in most medical schools. The plethora of new studies and the increasing questioning of medical decision-making point to the benefits of a better understanding by practitioners of the critical issues in clinical research design. Because of the high visibility and quantitative nature of cardiology, the importance of developing these skills among cardiologists is paramount.
History
The first chapter of the book of Daniel in the Bible describes a comparative experiment in which Daniel urges a servant to experiment with refraining from eating the king’s food, and, indeed, the healthier diet is associated with the finding that “at the end of ten days their countenances appeared fairer and they were fatter in flesh, than all the youths that did eat of the king’s food.” The modern human clinical trial, however, was not pioneered until 1747, when James Lind tried six different treatments for scurvy in 12 sailors; despite the small sample size, the virtues of oranges and lemons were evident (1). However, partly because of inadequate sample size, he failed to recognize the primary beneficial effect of oranges and lemons, considering “pure dry air” to be the most beneficial intervention.
In the nineteenth century P. C. A. Louis, in a treatise entitled La Methode Numerique, developed the concept of comparative observational studies (2,3). In casting doubt on the common practice of bloodletting, he clearly identified the concept of confounding, and he emphasized the need to have comparable groups when making comparisons. In the 1840s, Semmelweis (4) noted that maternal mortality was higher in women giving birth in hospitals in which deliveries were attended by physicians than in women giving birth in hospitals in which deliveries were attended by midwives. He succeeded in demonstrating that the excess mortality was related to contamination from autopsies and succeeded in separating autopsy teaching from attending childbirth by physicians, which led to a drop in maternal mortality.
The first randomization was performed by Fisher and Mackenzie in 1926 in an agricultural study (5). In developing analysis of variance, they recognized that experimental observations must be independent and not confounded to allow full acceptance of the statistical methodology. They therefore randomized different plots to different approaches to the application of fertilizer. Amberson is credited with the first randomization of patients, in a 1931 trial of tuberculosis therapy in 24 patients, using a coin toss to make treatment assignments (1). The British Medical Research Council trial of streptomycin in the treatment of tuberculosis began the modern era of clinical trials in 1948 (6). This trial established the principles of the use of randomization in large numbers of patients and set guidelines for administration of the experimental therapy and objective evaluation of outcomes.
Modern outcomes research was presaged by important observations from Florence Nightingale, who observed a higher mortality in hospitals than out of hospitals in the treatment of amputation. Her notes provide a detailed accounting of observational study methodology in the adjustment for differences in outcome as a function of baseline characteristics.
In 1914, Codman (7) wrote a treatise about measurement of outcomes that provides a classic discussion of the need for hospitals to evaluate outcomes. He proposed that reimbursement should be based on demonstration of results. Unfortunately, his message was not popular, and his hospital went bankrupt.
In the last decade, computers have enabled rapid accumulation of data in thousands of patients in studies conducted throughout the world. R. Peto, S. Yusuf, P. Sleight, and R. Collins developed the concept of the large, simple trial in ISIS-I (8), beginning with the concept that only by randomizing 10,000 patients could the beneficial effects of β-blockers be understood. The development of client server architecture provided a mechanism for aggregating large amounts of data and distributing them quickly to multiple users. Finally, the most recent advances in the development of the World Wide Web provide an opportunity to share information instantaneously in multiple locations around the world.
Phases of Evaluation of Therapies
Evaluating therapies and interpreting the results as they are presented requires an understanding of the goals of the investigation; these goals can be conveniently categorized in a similar manner to the nomenclature used by the U.S. Food and Drug Administration to characterize the phase of investigation in clinical trials (Table 43.1). Conclusions drawn from a small pilot study of a therapy never before given to humans should be quite different from conclusions drawn from a study measuring clinical outcomes comparing two therapies used in current practice; thus regulatory authorities have developed a nomenclature for discussing these phases of investigation.
When a new therapy is devised, it must first be studied in normal, healthy volunteers to ensure that no unexpected side effects will occur. This type of study is commonly referred to as Phase I. In some cases, when concern exists that the therapy may be highly toxic, patients at the advanced stage of disease (with presumably little to lose and much to gain if the treatment is successful) are evaluated instead. Many novel therapies affecting the coagulation system in which the risk of bleeding would not be acceptable for a normal volunteer fall into this category. The most extreme example of this approach was the use of recently deceased subjects (“neomorts”) in the initial studies of glycoprotein IIb/IIIa inhibitors (9). Although Phase I trials may generate substantial enthusiasm, they should not be used to make recommendations for routine clinical practice.
After the new therapy demonstrates some physiologic effect in the first phase of testing, the therapy must, in Phase II, demonstrate both desired physiologic activity and evidence of clinical benefit in the clinically relevant disease, at least from the perspective of “surrogate” pathophysiologic endpoints. For example, a heart failure therapy would need to demonstrate improvement in cardiac output, left ventricular function, or neurohormonal status. Often investigators are tempted to use these surrogate endpoints to recommend major changes in practice, but multiple experiences have demonstrated the danger of this approach to recommending changes in clinical practice (10). Often, even if the surrogate endpoints appear to be improved, large enough trials may show that the clinical outcomes are adversely affected. One of the major unresolved conceptual issues in modern cardiovascular research is how to use Phase II data to make wise choices about proceeding to the next phase.
The third phase, commonly referred to as the “pivotal” phase, evaluates the therapy in the relevant clinical context with the goal of determining whether the therapy should be used in clinical practice. For Phase III, the relevant endpoints include measures that can be recognized by patients as important: survival time, major clinical events, quality of life, and cost. A well-designed clinical trial with a positive effect on clinical outcomes justifies serious consideration for a change in clinical practice and certainly provides grounds for regulatory approval for sales and marketing.
After a therapy is in use and approved by regulatory authorities, Phase IV begins. Traditionally, Phase IV has been viewed as the monitoring of the use of a therapy in clinical practice, with a responsibility of developing more effective protocols for the use of that therapy, based on inference from observations and reporting of adverse events. The importance of this phase has evolved from the recognition that many circumstances
experienced in clinical practice will not have been encountered in randomized trials completed at the time of regulatory approval. Examples of Phase IV studies include the evaluation of new dosing regimens, as in several ongoing comparisons of low-dose versus high-dose angiotensin-converting-enzyme inhibition in patients with heart failure and the prospective registries of use of therapies such as the National Registry of Myocardial Infarction (NRMI) (11). Recently, with an increasing array of effective therapies, Phase IV is viewed as a time to compare one effective marketed therapy against another. In some cases this need arises because of changing doses or expanding indications for a therapy, whereas in other cases, the Phase III trials did not provide the relevant comparisons for a particular therapeutic comparison. GUSTO-I is an example of a Phase III trial in which a new dosing regimen of alteplase (accelerated dosing) was compared with a standard dose of streptokinase (12). The ongoing GUSTO-III Trial is an example of a Phase IV trial initiated because reteplase was approved by regulatory authorities based on a comparison with streptokinase, but it was not compared with accelerated alteplase; thus, GUSTO-III is performing that comparison because accelerated alteplase is the reference standard for practice in the United States.
experienced in clinical practice will not have been encountered in randomized trials completed at the time of regulatory approval. Examples of Phase IV studies include the evaluation of new dosing regimens, as in several ongoing comparisons of low-dose versus high-dose angiotensin-converting-enzyme inhibition in patients with heart failure and the prospective registries of use of therapies such as the National Registry of Myocardial Infarction (NRMI) (11). Recently, with an increasing array of effective therapies, Phase IV is viewed as a time to compare one effective marketed therapy against another. In some cases this need arises because of changing doses or expanding indications for a therapy, whereas in other cases, the Phase III trials did not provide the relevant comparisons for a particular therapeutic comparison. GUSTO-I is an example of a Phase III trial in which a new dosing regimen of alteplase (accelerated dosing) was compared with a standard dose of streptokinase (12). The ongoing GUSTO-III Trial is an example of a Phase IV trial initiated because reteplase was approved by regulatory authorities based on a comparison with streptokinase, but it was not compared with accelerated alteplase; thus, GUSTO-III is performing that comparison because accelerated alteplase is the reference standard for practice in the United States.
TABLE 43.1 Phases of Evaluation of New Therapies | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Critical General Concepts
With rare exceptions, the purpose of a Phase III or Phase IV clinical trial or outcome study is to estimate what is likely to happen to the next patient given one treatment or the other. To assess the degree to which the proposed study enhances the ability to understand what will happen to the next patient, the investigator must be aware of an array of methodologic and clinical issues. Although this task requires substantial expertise and experience, the issues can be considered in a broad framework. The broadest, but most essential concepts in understanding the relevance of a clinical study to practice are the concepts of validity and generalizability. Table 43.2 illustrates an approach to these issues, developed by the McMaster group, to be used when reading the literature.
TABLE 43.2 Questions to Ask When Reading and Interpreting the Results of a Clinical Trial | |
|---|---|
|
Validity
The most fundamental question about a clinical trial is whether the result is valid. Are the results of the trial internally consistent? Would the same result be obtained if the trial were repeated? Was the trial design adequate, including blinding, endpoint assessment, and statistical analyses? Of course, the most compelling evidence of validity in science is replication. If the results of a trial or study remain the same when the study is repeated, especially in a different clinical environment by different investigators, the results are likely to be valid.
Generalizability
Given a valid clinical trial result, it is equally important to determine whether the findings are generalizable. Unless the findings can be replicated and applied in multiple practice settings, little has been gained by the trial. Because it is impossible to replicate every clinical study in practice, it is especially important to understand the inclusion and exclusion criteria for patients entered into the study and to have an explicit understanding of additional therapies that the patients received. For example, studies done in “ideal” patients without comorbid conditions or young patients without severe illness can be misleading when the results are applied to clinical practice.
Expressing Clinical Trial Results
The manner in which the results of clinical research are reported can profoundly influence the perception of practitioners using the information to decide which therapies to use. A clinical trial will produce a different degree of enthusiasm about the therapy tested when the results are presented in the most favorable light, even when the exact results are provided in addition to the risk reduction so that the practitioner could reconstruct the results in different ways (13). Multiple studies (14,15) have demonstrated that physicians are much more likely to recommend prescribing a therapy when the results are presented as a relative risk reduction rather than an absolute difference in outcomes.
A recent example of a trial that has evoked considerable interest based on a predominant depiction of relative treatment effect is the 4S trial comparing simvastatin with placebo in patients with elevated low-density-lipoprotein (LDL) cholesterol and a history of ischemic heart disease (16). The results have been mostly depicted as a 30% reduction in overall mortality and a 42% reduction in coronary mortality. Rarely are the results presented in terms of absolute differences (approximately 0.6% reduction in mortality per year). This difference is highly significant because more than 4,000 patients were randomized; the cost-effectiveness data support use of simvastatin in this situation (18). In contrast, the Bypass Angioplasty Revascularization Investigation (BARI) found almost exactly the same difference between coronary artery bypass grafting and percutaneous revascularization, but with just more than 1,800 patients randomized, this difference was not statistically significant, and the conclusion was reached that no major differences in outcome exist between the two technologies in patients with suitable coronary anatomy for either. If the same result had been achieved with 4,000 patients randomized, would the same conclusion have been drawn as for simvastatin?
The results of pragmatic clinical trials are most appropriately expressed in terms of the number of poor outcomes prevented by the more effective treatment per 100 or 1,000 patients treated. This measure, representing the absolute benefit of therapy, number needed to treat (NNT), translates the results for specific populations studied into public health terms by quantifying how many patients would need to be treated to create a specific health benefit. The absolute difference can be used to assess quantitative interactions, that is, significant differences in the number of patients needed to treat to achieve a degree of benefit with a therapy as a function of the type of patient treated. An example is the use of thrombolytic therapy; the Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group demonstrated that 37 lives per 1,000 patients treated are saved when thrombolytic therapy is used in patients with anterior ST-segment elevation, whereas only 8 lives per 1,000 are saved when patients with inferior ST-segment elevation are treated (Fig. 43.1) (19). The direction of the treatment effect is the same, but the magnitude of the effect is different.
This approach becomes more complex with endpoints that are not discrete, such as exercise time. One approach to expressing trial results when the endpoint is a continuous measurement is to define the minimal clinically important difference (the smallest difference that would lead practitioners to change their practices) and to express the results in terms of the NNT to achieve that minimal clinically important difference.
The relative benefit of therapy, on the other hand, is the best measure of the treatment effect in biologic terms. This concept is defined as the proportional reduction in risk resulting from the more effective treatment and is generally expressed in terms of an odds ratio or relative risk reduction. The relative treatment effect can be used to assess qualitative interactions, which represent statistically significant differences in the direction of the treatment effect as a function of the type of patient treated. In the FTT analysis (19), the treatment effect in patients without ST-segment elevation is heterogeneous compared with patients with ST-segment elevation. Figure 43.2 gives the calculations for commonly used measures of treatment effect. A particularly useful display of data is the odds ratio plot, as shown in Figure 43.3. Both absolute and relative differences in outcome should be expressed in terms of point estimates and confidence intervals. This type of display gives the reader a balanced perspective because both the relative and absolute differences are important as well as the confidence in the estimate. Without the confidence intervals, the reader has difficulty knowing the precision of the estimate of the treatment effect.
Concepts Underlying Trial Design
As experience with multiple clinical trials is accumulating, some general concepts seem worth emphasizing. These generalities do not always pertain, but they serve as useful guides to the design or interpretation of trials.
Treatment Effects Are Modest
The most common mistake in designing clinical trials is overestimation of the expected treatment effect. Most individuals heavily involved in therapeutic development cannot resist the assumption that the pathway being targeted is the most important contributor to patient outcome. Unfortunately, relative reductions in adverse clinical outcomes exceeding 25% are extremely uncommon.
When treatments affecting outcome in acute myocardial infarction have been assessed, small trials typically greatly overestimate the effect observed in subsequent larger trials. The reasons for this observation are not entirely clear. One important factor is negative publication bias. Of the many small studies done, the positive ones tend to be published. A second factor
could be analogous to regression to the mean in observational studies: when a variety of small trials are done, only those with a substantial treatment effect are likely to be continued into larger trials. Of course, in most cases the small trials have so much uncertainty in the estimate of the treatment effect that the true effect of many of the promising therapies is overestimated, whereas the effect of some of the therapies showing little promise based on point estimates from small studies is underestimated. Thus, when larger studies are completed, giving a more reliable estimate of treatment effect, the estimate of benefit tends to regress toward average.
could be analogous to regression to the mean in observational studies: when a variety of small trials are done, only those with a substantial treatment effect are likely to be continued into larger trials. Of course, in most cases the small trials have so much uncertainty in the estimate of the treatment effect that the true effect of many of the promising therapies is overestimated, whereas the effect of some of the therapies showing little promise based on point estimates from small studies is underestimated. Thus, when larger studies are completed, giving a more reliable estimate of treatment effect, the estimate of benefit tends to regress toward average.
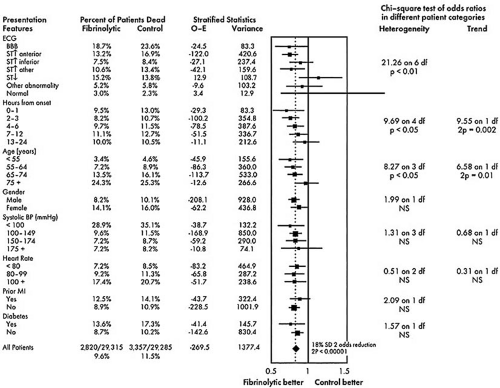 FIGURE 43.1. Proportional effects of fibrinolytic therapy on mortality. BBB, bundle branch block; BP, blood pressure; df, degrees of freedom; ECG, electrocardiography; MI, myocardial infarction; NS, not significant; O-E, observed–expected; SD, standard deviation. |
Qualitative Interactions Are Uncommon
A reversal of treatment effect as a function of baseline characteristics is unusual. Many training programs have taught clinicians that many therapies are effective only in very select subsets of the population, yet there are few examples demonstrating such a targeted effect. One exception in cardiovascular trials is the probable harm of thrombolytic therapy for patients without ST-segment elevation, although this finding lacks adequate statistical power, and the recently published LATE Trial found that thrombolytic therapy was beneficial in these patients (20).
Quantitative Interactions Are Common
When therapies are beneficial for specific patients with a given diagnosis, they are generally beneficial to most patients with that diagnosis. However, therapies commonly provide a differential absolute benefit as a function of the severity of the patient’s illness. Given the same relative treatment effect, the number of lives saved or events prevented will be greater when the therapy is applied to patients with a greater underlying risk. Examples of this concept include the greater benefit of angiotensin-converting-enzyme inhibitors in patients with markedly diminished left ventricular function, the larger benefit of thrombolytic therapy in patients with anterior infarction, and the greater benefit of bypass surgery in older patients than in younger patients.
Unintended Targets Are Common
Therapies are appropriately developed by finding a pathophysiologic pathway or target that can be altered and by exploiting that concept using a model that does not involve the intact human. Despite all good intentions, proposed therapies
frequently either work via a different mechanism than the one for which the therapy was devised or affect an entirely different system. Examples include thrombolytic therapy for myocardial infarction, which was developed using coronary thrombosis models; unfortunately, this therapy also affects the intracranial vessels. Inotropic therapies for heart failure were developed using measures of cardiac function, but many of these agents, which clearly improve cardiac function acutely, also cause an increase in mortality, perhaps due to a detrimental effect on the neurohormonal system. Hormone replacement therapy treats hot flashes and increases bone density and its “beneficial effects” on lowering LDL cholesterol have been documented multiple times. Unfortunately, after the large clinical trials were done, it became clear that offsetting effects, including an elevation of C-reactive protein, offset these beneficial effects, giving a net effect of an increase in cardiovascular events (21,22). Perhaps no case has received more attention recently than the case of the coxibs, which were developed based on biologic demonstration that selectively inhibiting the COX-2 enzyme could reduce inflammation with a lower risk of gastric erosion. Again, unfortunately the net effect on human biology appears to have caused an increase in cardiovascular events (23,24).
frequently either work via a different mechanism than the one for which the therapy was devised or affect an entirely different system. Examples include thrombolytic therapy for myocardial infarction, which was developed using coronary thrombosis models; unfortunately, this therapy also affects the intracranial vessels. Inotropic therapies for heart failure were developed using measures of cardiac function, but many of these agents, which clearly improve cardiac function acutely, also cause an increase in mortality, perhaps due to a detrimental effect on the neurohormonal system. Hormone replacement therapy treats hot flashes and increases bone density and its “beneficial effects” on lowering LDL cholesterol have been documented multiple times. Unfortunately, after the large clinical trials were done, it became clear that offsetting effects, including an elevation of C-reactive protein, offset these beneficial effects, giving a net effect of an increase in cardiovascular events (21,22). Perhaps no case has received more attention recently than the case of the coxibs, which were developed based on biologic demonstration that selectively inhibiting the COX-2 enzyme could reduce inflammation with a lower risk of gastric erosion. Again, unfortunately the net effect on human biology appears to have caused an increase in cardiovascular events (23,24).
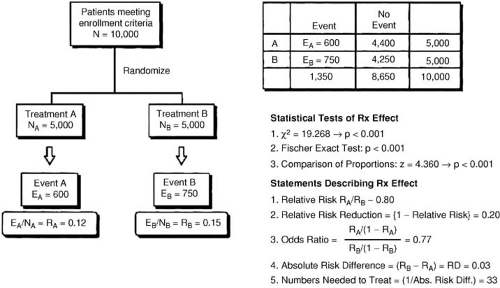 FIGURE 43.2. Summary measures of treatment effect. Rx, therapy. |
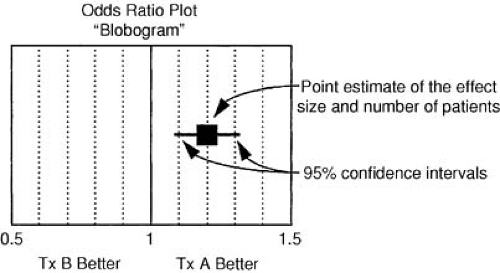 FIGURE 43.3. Odds ratio plot. Tx, treatment. |
General Design Considerations
When reading the results of a clinical study or designing a study, the purpose of the investigation is critical to placing the outcome in context. Those who design the investigation have the responsibility of constructing the project and presenting the results in a manner reflecting the intent of the study. In a small Phase II study, an improvement in a surrogate pathophysiologic outcome is exciting and could easily lead the investigator to overstate the clinical implications of the finding. Similarly, megatrials with little data collection seldom give useful information about disease mechanisms unless carefully planned substudies are performed. The structural characteristics of trials can be characterized as a function of the following attributes.
Pragmatic versus Explanatory
Most clinical trials are designed to demonstrate a physiologic principle as part of a chain of causality of a particular disease. Such trials, termed explanatory trials, need only be large enough to prove or disprove the hypothesis being tested. Major problems have arisen because of the tendency of those doing explanatory trials to generalize the findings into recommendations about clinical therapeutics.
Trials designed to answer questions about which therapies should be used are called pragmatic trials. These trials should have clinical outcomes as the primary endpoint so that when the trial is complete the result will inform the practitioner and the public whether using the therapy in the manner tested will result in better clinical outcomes than the alternative approaches. These trials generally require much larger sample sizes and a more heterogeneous population.
The decision about whether to perform an explanatory trial or a pragmatic trial has major implications for the design of the study. When the study is published, the reader must also take into account the intent of the investigators because the implications for practice or knowledge will vary considerably depending on the type of study.
Entry Criteria
In an explanatory trial, the entry criteria should be carefully controlled so that the particular measurement of interest will not be confounded. For example, a trial designed to determine whether a treatment for heart failure improves cardiac output should study patients who are stable enough for elective hemodynamic monitoring. In contrast, in a pragmatic trial, the general goal is to include patients who represent the population seen in clinical practice and whom the organizers of the study believe can make a plausible case for benefit in outcome. From this perspective, the number of entry and exclusion criteria should be minimized because the rate of enrollment will be inversely proportional to the number of criteria.
Data Collection Form
The data collection form provides the information on which the results of the trial are built; if an item is not included on the data collection form, it will obviously not be available at the end of the trial. On the other hand, the likelihood of collecting accurate information is inversely proportional to the amount of data collected. In an explanatory trial, patient enrollment is generally not an issue because small sample sizes are needed. However, in a pragmatic trial there is almost always an imperative to enroll patients as quickly as possible. Thus, a fundamental concept in pragmatic trials is to keep the data collection form as brief as possible.
The ultimate example of this approach is the ISIS approach of collecting only the front of a single page in a clinical trial (8). This approach has allowed the enrollment of tens of thousands of patients in mortality trials with no reimbursement to the health care providers enrolling patients. Regardless of the ultimate length of the data collection instrument, it is critical to include only information that will be useful in analyzing the trial outcome.
Ancillary Therapy
Decisions about the use of nonstudy therapies in a clinical trial are critical to its validity and generalizability. Including therapies that will interact in a negative way with the experimental agent could ruin the chance to detect a clinically important treatment advance. Especially in a physiologic experiment, interfering with the primary question with another therapy would be a serious problem.
Alternatively, in a pragmatic trial the goal is to evaluate the therapy in the context in which it will be used. Because clinical practice is not managed by a prespecified algorithm, evaluation of the experimental therapy in the context of such an approach is likely to give an unrealistic approximation of the likely impact of the therapy in clinical practice. When the pragmatic approach is taken, however, those who manage and analyze the trial must be aware of the issue of “intensification.” This term refers to the phenomenon in which therapy is more aggressive in the arm randomized to the less effective treatment, thereby equalizing the outcomes in the two arms of the trial. The recently reported FIELD Trial suffered from this problem (25).
Multiple Randomizations
Until recently, enrolling a patient in multiple simultaneous clinical trials was considered to be ethically questionable. The origin of this ethical concern is unclear, but it seems to have arisen from a general impression that clinical research encroaches on clinical practice, and thus more clinical research would be detrimental. More recently, the concept has been proposed that when the best treatment is not known, randomization is desirable (the uncertainty principle). Stimulated by the apparent need to develop multiple therapies simultaneously in the treatment of acquired immunodeficiency syndrome, the concept of multiple randomizations has been reconsidered.
Factorial trial designs represent a specific approach to multiple randomizations with advantages from the statistical and the clinical perspectives. Because most patients are now treated with multiple therapies, the factorial design represents a clear method for determining whether therapies add to each other, work synergistically, or nullify the effects of one or both therapies being tested. As long as a significant interaction does not exist between the two therapies being tested, both can be tested in a factorial design with a sample size close to the size needed to test one therapy.
Legal and Ethical Issues
Multiple considerations pertain to the question of whether a clinical trial should be done and how the results of the trial should be disseminated. Perhaps too little thought is typically given to the critical ethical issues before trials are done. Three general principles should be considered when clinical investigation is considered (26). Respect for persons embodies the concept that the investigator recognizes that patients are not simply sources of data, but that they are individuals deserving of the opportunity to provide informed consent, and that they should be considered as collaborators in the investigation. Beneficence refers to the concept that the research will be done to improve the state of knowledge and that the risks assumed by the participants in the study are not out of proportion to the potential benefits. This concept has particular relevance to failure to publish negative clinical trials; when a patient has consented to participation in an experiment, failure to use the information gained to advance the state of knowledge raises a concern. Justice refers to the concept that the benefits, risks, and inconvenience should be distributed fairly; because volunteering for a study implies that a risk is taken to benefit others, efforts must be made to avoid subjecting particular groups to risk so as to benefit other particular groups.
Medical Justification
Each of the proposed treatments in the trial must be within the realm of currently acceptable medical practice for the specific medical condition of the patient. Difficulties with consideration of medical justification typically arise in two areas: studies are generally initiated because there is a reason to believe that one therapeutic approach is better than another, and many currently accepted therapies have never been subjected to the type of scrutiny being applied to new therapies. These factors create a dilemma for the practitioner, who may be uncomfortable with protocols asking for a change in standard practice. An approach to this dilemma called the “uncertainty principle” has been proposed. In this scheme, the clinician should be comfortable offering entry into a study at any time he or she is substantially uncertain about whether one of the treatments is in fact better than the other. The patient, of course, is given the opportunity to review the situation and make a decision,
but for most patients the physician’s recommendation will be a critical factor in deciding whether to participate in a study.
but for most patients the physician’s recommendation will be a critical factor in deciding whether to participate in a study.
Groups of Patients versus Individuals
The ethical balance typically depends on the good of larger numbers of patients versus the good of individuals involved in the trial. Examples are accumulating in which a therapy appeared to be better based on preliminary results or small studies and then was shown to be inferior based on adequately sized studies. These experiences have led some authorities to argue that clinical practice should not change until a highly statistically significant difference in outcome is demonstrated (27). Indeed, the standard for acceptance of a drug for labeling by the cardiorenal group at the U.S. Food and Drug Administration is two adequate and well-controlled trials, each independently reaching statistical significance. If the alpha for each trial is .05, an alpha value of .0025 (.05 times .05) would be needed for both to be positive. The counterargument is that the physician advising the individual patient should let the patient know which treatment is most likely to lead to the best outcome. In fact, Bayesian calculations could be used to provide running estimates of the likelihood that one treatment is better than the other.
Informed Consent
The U.S. Department of Health and Human Services and the International Guidelines for Biomedical Research Involving Human Subjects provide standards for the process of informed consent. Elements common to informed consent have been defined and should pertain regardless of the exact nature of the investigation. The research project, including the names of the investigators, should be described to the patient. The procedures that will be involved, including follow-up information, should be described in enough detail that the patient can envision what participation will entail. The potential risks and benefits of participation should be described, including a description of alternatives to participation in the study. The patient should be reassured that participation in the study is voluntary and that the confidentiality of the patient will be protected. Finally, the patient should be given the chance to ask questions.
An increasing area of confusion is the distinction between clinical investigation and measures taken to improve the quality of care as an administrative matter. The argument has been made that the former requires individual patient informed consent, whereas the latter falls under the purview of the process of medical care and does not require individual consent.
Several special situations must be considered in cardiovascular studies. In a cardiac emergency, there is often insufficient time to explain the research project in exacting detail and obtain informed consent. When therapies for treatment of acute myocardial infarction are compared, the time to administration of therapy is a critical determinant of outcome, and time spent considering participation in a protocol could increase the risk of death. Accordingly, an abbreviated consent to participate followed by a more detailed explanation later in the hospitalization has been sanctioned. An even more complex situation occurs in research concerning treatment of cardiac arrest. Clinical investigation in this field almost came to a halt because of the impossibility of obtaining informed consent. After considerable national debate, such research is now being done only after careful consideration by the community of providers and citizens about the potential merits of the proposed research.



