Incidence and Etiology
The incidence of congenital heart disease in the United States is approximately 8 per 1000 live births.1,2 Many infants who are born alive with cardiac defects have anomalies that do not represent a threat to life, at least during infancy. Almost one-third of those infants, or 2.6 per 1000 live births, however, have critical disease, which is defined as a malformation severe enough to result in cardiac catheterization, cardiac surgery, or death within the first year of life.3 Today, with early detection and proper management, the majority of infants with critical disease can be expected to survive the first year of life. Most who now survive infancy will join the increasingly large cohort of adults with congenital heart disease.
Estimates of the incidence of specific lesions vary, depending on whether the data are drawn from infants or older children and whether the diagnosis is based on clinical, echocardiography, catheterization, surgical, or postmortem studies.1-4 The incidence in other countries is remarkably similar to that reported for the United States.5,6 Despite these differences in case material, except for bicuspid aortic valve and mitral valve prolapse, it is apparent that ventricular septal defect (VSD) is the most common malformation, occurring in 28% of all patients with congenital heart disease (Table 83–1).
| Defect | Percentage of Casesa (Averaged) |
|---|---|
| Ventricular septal defect (VSD) | 28.3 |
| Pulmonary stenosis | 9.5 |
| Patent ductus arteriosus | 8.7 |
| VSD with pulmonary stenosisb | 6.8 |
| Atrial septal defect, secundum | 6.7 |
| Aortic stenosis | 4.5 |
| Coarctation of aorta | 4.2 |
| Atrioventricular canalc | 3.5 |
| Transposition of the great arteries | 3.4 |
| Aortic atresia | 2.4 |
| Truncus arteriosus | 1.6 |
| Tricuspid atresia | 1.2 |
| Anomalous pulmonary venous connection | 1.1 |
| Double-outlet right ventricle | 0.8 |
| Pulmonary atresia without VSD | 0.3 |
Among 2251 infants with critical congenital heart disease in the New England Regional Infant Cardiac Program,3 53.7% were male. Certain defects, however, are more much common in one sex than in the other. Aortic stenosis occurs more often in boys (4:1), and atrial septal defects (ASDs) occur more frequently in girls (2.5:1).
Although earlier theories concerning the etiology of congenital heart diseases suggested that most defects were multifactorial—that is, the malformations are caused by a combination of a hereditary predisposition (presumably caused by abnormalities in the genetic code) and an environmental trigger7—more recent advances in molecular biology suggest that a much higher percentage are caused by point mutations.8
Some abnormalities are caused by chromosomal aberrations (see Chap. 82). Trisomy 21 (Down syndrome) is highly associated with complete atrioventricular (AV) canal, VSDs, and tetralogy of Fallot, and children with Turner syndrome (XO chromosome) frequently have coarctation of the aorta. Other anomalies are caused by teratogens: VSD in fetal alcohol syndrome, Ebstein anomaly in a fetus with prenatal exposure to lithium, and patent ductus arteriosus (PDA) in mothers who contracted rubella during the first trimester are examples.
Some syndromes are inherited as single-gene defects and have congenital heart disease as one of their manifestations. Holt-Oram syndrome, an association of radial limb abnormalities and ASDs, is caused by an abnormality of a T-box transcription factor Tbx5. DiGeorge syndrome (also known as velocardiofacial syndrome), an association of villous insufficiency, abnormalities of the thymus and parathyroid glands, and characteristic facial features, is associated with abnormalities of the conotruncus, resulting in a high proportion of infants born with tetralogy of Fallot, truncus arteriosus, or interrupted aortic arch. Deletions of chromosomes 22 (22q11) and 10 (10p13) have both been demonstrated to result in clinical DiGeorge syndrome.9 Williams syndrome, characterized by hypercalcemia, cognitive deficits, elfin facies, and supravalvar aortic and pulmonary stenosis, often results from a microdeletion at chromosome 7q11.23; the elastin gene is a component of the typical deletion.10 Nonsyndromic single-gene defects causing congenital heart defects have also recently been described. For example, mutations in the transcription factor NKX2.5 have been demonstrated in kindreds with atrial septal defects and atrioventricular conduction delay.11 It is clear now that a higher proportion of congenital heart disease than previously thought is caused by single-gene defects and that the same malformation may be caused by mutant genes at different loci.8,12 Given the variability in penetrance and expressivity noted even in patients with identical genetic alterations, many cases of congenital heart defects are most likely the result of multiple genetic alterations that, interacting with environmental factors, increase susceptibility to congenital heart disease. With increasing knowledge of molecular mechanisms, it seems inevitable that the etiology and pathogenesis of the many forms of congenital heart disease will be clarified increasingly in the years ahead.
Fetal Circulation and the Transition to Neonatal and Adult Circulation
The fetus obtains all metabolic necessities, including oxygen, from the placenta. The fetal circulation allows most of the right ventricular output to bypass the lungs and instead to perfuse the placenta. Most of the understanding of this adaptation comes from more than 40 years of research,13-21 primarily on fetal lambs. The fetal circulation is arranged in parallel fashion rather than in series, with mixing at the atrial (foramen ovale) and great vessel (ductus arteriosus) levels (Fig. 83–1), which allows for shunting of blood to provide more oxygenated blood to the developing brain and deoxygenated blood to return to the placenta. Normally, systemic venous blood enters the right atrium via the superior or inferior vena cava. From the inferior vena cava, blood is diverted by the crista dividens through the foramen ovale into the left atrium, so that approximately 27% of combined ventricular output reaches the left ventricle, with the remainder passing through the tricuspid valve to the right ventricle. This left atrial flow mixes with a small volume of pulmonary venous return to enter the left ventricle and the ascending aorta. Most of this output perfuses the coronary arteries, head, and upper body vessels, with a small proportion crossing the aortic arch to the descending aorta. Right ventricular output enters the main pulmonary artery, where approximately 90% (59% of combined ventricular output) is diverted through the ductus arteriosus to the descending aorta. Thus approximately two-thirds of the combined cardiac output passes through the right side of the heart, and one-third passes through the left side of the heart.
Figure 83–1.
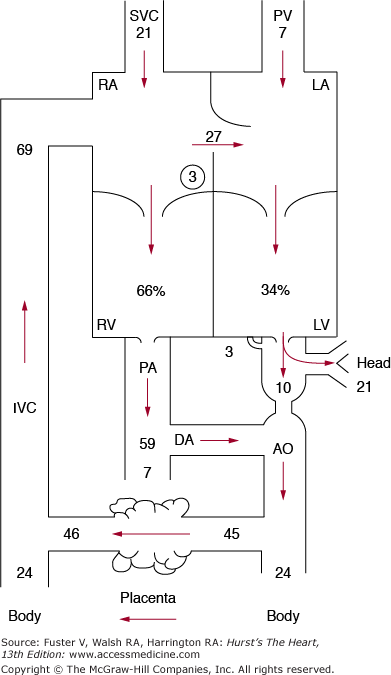
The course of the circulation in a late-gestation fetal lamb. The numbers represent the percentage of combined ventricular output. Some of the return from the inferior vena cava (IVC) is diverted by the crista dividens in the right atrium (RA) through the foramen ovale into the left atrium (LA), where it meets the pulmonary venous return (PV), passes into the left ventricle (LV), and is pumped into the ascending aorta. Most of the ascending aortic flow goes to the coronary, subclavian, and carotid arteries, with only 10% of combined ventricular output passing through the aortic arch (indicated by the narrowed point in the aorta) into the descending aorta (AO). The remainder of the inferior vena cava flow mixes with the return from the superior vena cava (SVC) and coronary veins, passes into the right atrium and right ventricle (RV), and is pumped into the pulmonary artery (PA). Because of the high pulmonary resistance, only 7% passes through the lungs (PV), with the rest going into the ductus arteriosus (DA) and then to the descending aorta (AO), the placenta, and the lower half of the body. Reproduced with permission from Freed MD. Fetal and transitional circulation. In: Keane et al.5
The oxygen saturation of fetal blood is considerably lower than that in a newborn or infant because of the placenta’s less efficient oxygen exchange compared with that the lungs (Fig. 83–2). The blood with the highest saturation (approximately 70%) is that returning from the placenta. As described above, some of this higher-saturation blood is diverted across the foramen ovale, so that saturation on the left side of the heart (65%) is somewhat higher than it is on the right side (55%), preferentially supplying the developing fetal brain with the highest saturation blood possible. The lower-saturation blood (some 55%) on the right side of the heart passes preferentially through the ductus arteriosus to the placenta, thus increasing the efficiency of oxygen pickup. The presence of high levels of fetal hemoglobin with a greater than normal affinity for oxygen hemoglobin promotes more efficient placental oxygen exchange related to a leftward shift of the oxygen dissociation curve.
Figure 83–2.
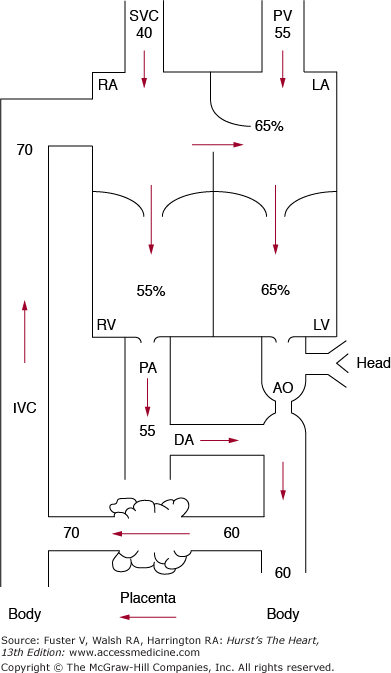
The numbers indicate the percent of oxygen saturation in a late-gestation lamb. The oxygen saturation is highest in the inferior vena cava, representing that primarily from the placenta. The saturation of blood in the heart is slightly higher on the left side than on the right side. The abbreviations in this diagram are the same as those in Fig. 83–1. Reproduced with permission from Freed MD. Fetal and transitional circulation. In: Keane et al.5
The wide communication at the atrial level (foramen ovale) allows for near equalization of atrial and ventricular end-diastolic pressures. Similarly, at the great vessel level, the nonrestrictive ductus arteriosus allows equalization of systolic pressures in the aorta and the pulmonary artery and, in the absence of aortic or pulmonic stenosis, at the ventricular level (Fig. 83–3).
Figure 83–3.
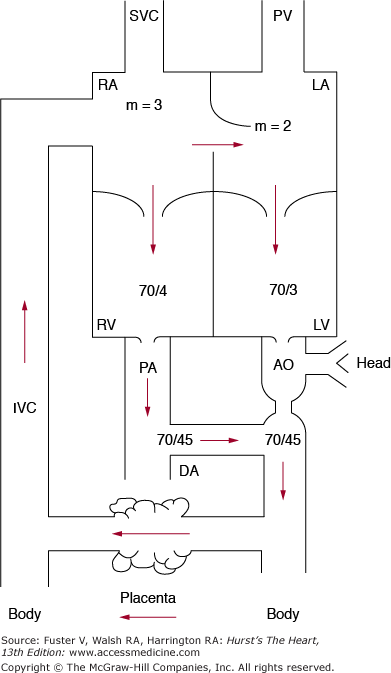
The numbers indicate the pressures observed in late-gestation lambs. Because large communications between the atrium and the great vessels are present, the pressures on both sides of the heart are virtually identical. The abbreviations are the same as those in Fig. 83–1. Reproduced with permission from Freed MD. Fetal and transitional circulation. In: Keane et al.5
Within a few moments after birth, the circulatory physiology must switch rapidly from the placenta to the lung as the target organ for oxygen exchange. Failure of any one of a number of a complex series of pulmonary and cardiac events may result in cerebral or generalized hypoxemia, with lasting damage or death. With the onset of spontaneous respiration, the lungs expand and the pulmonary arterioles, which have been vasoconstricted, dilate. The acute reduction in pulmonary vascular resistance results from both simple physical expansion of the lung with the onset of respiration and the vasodilation of the pulmonary resistance vessels, probably partly as a result of the high level of oxygen in alveolar gas. Simultaneously, the placenta is removed from the circulation either by clamping the umbilical cord or by constriction of the umbilical arteries. This sudden increase in systemic vascular resistance and drop in pulmonary vascular resistance causes blood leaving the right ventricle to enter the lung rather than the ductus arteriosus. The subsequent increase in pulmonary venous return to the left atrium increases left ventricular end-diastolic and left atrial pressure, shutting the flap valve of the foramen ovale against the edge of the cristae dividens, reversing the right-to-left atrial level shunt. In the presence of a lower pulmonary vascular resistance than systemic vascular resistance, some left-to-right (aorta to pulmonary artery) shunting occurs through the ductus arteriosus. The mechanism for closure of the ductus arteriosus is not completely understood. The increased level of oxygen probably causes vasoconstriction of the ductal musculature, but there are strong suggestions that a reduction in circulating prostaglandins of the E series plays a role. Typically, within 3 or 4 days, the vasoconstrictive ductal closure becomes irreversible, when cellular necrosis of the endothelium leads to obliteration of the lumen. The pulmonary artery pressure drops to approximately half systemic levels within a day or so but requires another 2 to 6 weeks to decrease to adult levels.
The structure and hemodynamics of the fetal circulation have significant consequences in a neonate with congenital heart disease.22 The parallel circulation with connections at the atrial and great vessel level allows a wide variety of congenital cardiac malformations to exist in utero while still maintaining placental oxygen exchange and tissue delivery. For example, atresia of the tricuspid or mitral valve, although devastating after birth, does not have a significant effect in utero. Furthermore, because the right ventricle performs two-thirds of the cardiac work before birth, the left ventricle is underloaded, possibly explaining why heart failure is not uncommonly seen with congenital defects. Because the normal flow across the aortic isthmus is relatively low (only approximately 10% of combined ventricular output), this area is especially vulnerable to small changes in flow. A somewhat small foramen ovale may result in left-sided hypoplasia, which is almost always associated with narrowing (coarctation) or atresia (interruption) at the distal transverse aorta just proximal to ductal insertion. Other congenital heart defects that subtly alter the amount of flow across the aortic isthmus, such as subaortic stenosis or bicommissural aortic valve, can also result in postnatal development of coarctation of the aorta.
Because the pulmonary blood flow in utero is less than 10% of combined ventricular output and increases four to five times at birth, anomalies that obstruct pulmonary venous return (such as cor triatriatum) may be masked in utero under conditions of low pulmonary blood flow. Finally, the low circulating levels of oxygen before birth (PO2 26-38 mm Hg and saturation at 50%-60%) may account for the relative level of comfort observed in infants with cyanotic heart disease. These infants may do well, at least in the short run, with a PO2 of 30 mm Hg and an aortic saturation of 50%—levels that would lead to cerebral and cardiac anoxia, acidosis, and death within a few minutes in an older child or adult.
Persistence of the fetal circulation, now more precisely called persistent pulmonary hypertension of the newborn, occurs when the pulmonary vascular resistance fails to decrease after birth. This results in high right ventricular and pulmonary artery pressure and right-to-left shunting through the patent foramen ovale and/or PDA. It occurs most commonly in full-term infants, with common etiologies including meconium aspiration syndrome, primary pulmonary hypertension of the newborn, respiratory distress syndrome, pneumonia, and sepsis.23 The precise cause of primary pulmonary hypertension of the newborn is not well understood, although prenatal constriction of the ductus arteriosus (often due to maternal intake of nonsteroidal anti-inflammatory medications) has clearly been implicated.24 Severe hypoxia usually is manifest in the first few hours of life with tachypnea and metabolic acidosis, and a chest roentgenogram shows diminished pulmonary vascular flow. With primary pulmonary hypertension of the newborn, no pulmonary parenchymal disease is evident. Physical examination may reveal a parasternal heave, a loud second heart sound, and a systolic murmur.
Polycythemia, transient myocardial ischemia from hypoglycemia, and cyanotic congenital cardiac defects must be excluded. A higher oxygen level in the right radial artery than a subdiaphragmatic site (most easily noted by cutaneous oximetry in the right arm and a lower extremity) confirms right-to-left shunting through the ductus arteriosus. In this setting, echocardiography is of the utmost importance to rule out structural heart disease, especially total anomalous pulmonary venous connection. The initial treatment includes an increase in the inspired oxygen level to restore normal partial pressure of oxygen, the correction of acidosis to a normal pH through ventilatory strategies that minimize barotrauma, surfactant administration to assist with lung alveoli recruitment, the reduction of pulmonary vascular resistance with inhaled nitric oxide, and inotropic support as necessary to support normal cardiac output.23,25 Treatment of severe disease with extracorporeal membrane oxygenation has proven lifesaving in a significant number of patients,26 although this therapy is now less frequently required with the recent availability of inhaled nitric oxide and newer ventilation strategies.
Complications of Congenital Heart Disease
Heart failure is a potentially lethal complication of congenital heart disease and occurs in more than 80% of infants who have malformations severe enough to require cardiac catheterization or surgery within the first year of life.27 The onset is usually within the first 6 months of life; it is rarely found after 1 year of age without a serious intercurrent problem such as infective endocarditis, pneumonia, or anemia.
Heart failure within the first 12 to 18 hours of life is usually a result of malformations that involve pressure or volume overload independent of pulmonary flow, as occurs with severe valvular regurgitation or a systemic arteriovenous fistula. Rarely, myocarditis may produce failure from the time of birth, as may congenital complete heart block or supraventricular tachycardia. Other causes in this age group include primary cardiomyopathy, severe polycythemia, anemia, and depressed myocardial contractility resulting from neonatal asphyxia, hypocalcemia, hypoglycemia, or sepsis. A majority of full-term infants presenting with severe heart failure during the remainder of the first week have critical obstruction to systemic arterial flow, which, in virtually all cases, is unmasked by narrowing or closure of the ductus arteriosus. Examples are aortic atresia, coarctation of the aorta, interruption of the aortic arch, and critical aortic stenosis. During the second week of life, aortic atresia and coarctation remain the most common causes of heart failure, but left ventricular volume overload from VSD, transposition of the great arteries with a VSD, and truncus arteriosus make their appearance. These malformations present as the pulmonary vascular resistance falls, increasing the left-to-right shunt. Statistically, VSD is the most frequent cause of congestive failure, followed by transposition, coarctation, complete AV canal, and PDA.
The most common symptom of heart failure is tachypnea with grunting, gasping breathing, or breathlessness with feeding. Observation of an undisturbed infant reveals nasal flaring and subcostal or intercostal retractions. A respiratory rate consistently above 60 is to be expected, and rates in the range of 90 to 100 are not uncommon. Poor weight gain from diminished intake and increased work of breathing is the rule. Cool, moist skin; a diminished and rapid arterial pulse; and hepatic enlargement are common accompanying signs. A gallop rhythm, pulmonary rales, and wheezing may be present. It may be difficult to distinguish the pulmonary findings of heart failure from those of pneumonia or bronchiolitis; indeed, many infants develop heart failure during an intercurrent pulmonary infection. Edema, if present, is usually found in the periorbital area and on the dorsa of the feet and hands. Cardiac enlargement is confirmed by chest roentgenography. Infants with malformations such as coarctation of the aorta and total anomalous pulmonary venous connection—abnormalities that usually are not characterized by an impressive murmur—are sometimes referred only after weeks of tachypnea and failure to thrive, when a chest x-ray taken to explore the possibility of lung disease shows cardiac enlargement.
When a sizable systemic-to-pulmonary communication exists in a premature infant, usually as a result of a PDA, signs of heart failure are often associated with signs of ventilatory failure.
Table 83–3 lists the sequelae of hypoxemia. Cyanosis, a bluish tinge to the color of the skin caused by the presence of at least 3 to 5 g/dL of reduced hemoglobin, is frequently the initial sign of congenital heart disease in an infant. It may also be an early sign of pulmonary, central nervous system, metabolic disease, or methemoglobinemia. Prompt distinction between cardiac and noncardiac cyanosis, usually by echocardiography, is extremely important, as palliation with prostaglandin E1 (PGE1) infusion followed by early surgical intervention has improved survival.
Hypoxia leading to cyanosis in congenital heart disease may be caused by heart failure with pulmonary edema and pulmonary venous desaturation and/or intracardiac right-to-left shunting. The hypoxia that is either a result of heart failure or of lung disease with ventilation–perfusion mismatch usually responds dramatically to oxygen administration, whereas hypoxia that is caused by cyanotic defects does not. Because many infants are relatively anemic during the first few months of life (hemoglobin concentration of 10.4-12 g/dL), cyanosis may be subtle, despite significant decreases in arterial PO2. When cyanosis has been present in older children for several months, the distal tips of the fingers and toes become hyperemic. Eventually, the capillary end loop dilation causes clubbing of the fingers and toes with a loss of the normal angle of the base of the nail and fingers. Also, with long-standing hypoxemia, the production of red blood cells increases to maintain the oxygen-carrying capacity of the blood (polycythemia). The increased hemoglobin concentration at any given oxygen saturation will result in more reduced hemoglobin, thus exaggerating the cyanosis.
The central nervous system may be the target organ of cerebrovascular accidents or brain abscess. Brain abscess is probably caused by bacteremia, primarily with mouth organisms that cross from the venous system to the arterial system from right-to-left shunting. The incidence seems to be directly related to arterial saturation and occurs mostly in older children and adolescents.28
Cerebrovascular accidents are directly caused by hypoxemia or indirectly in children who are polycythemic presumably secondary to sludging.29 The former group usually consists of infants younger than 2 years of age who are anemic and thus may have markedly reduced oxygen levels. The latter group consists of children or young adults who are polycythemic and have sludging or in situ microthrombosis. Interestingly, because iron deficiency leads to decreased deformity of red cells, sludging may occur with modest levels of polycythemia (hematocrit 55%-60%) in the presence of iron deficiency. With hematocrits in the range of 65% or higher, increased viscosity may lead to a cerebrovascular accident. Maintaining a proper level of hemoglobin has a salutary effect on hemodynamics and oxygen delivery in the presence of significant hypoxemia.30,31 Other systems may also be affected by hypoxemia or polycythemia. In older adolescents, the increase in hemoglobin breakdown may result in hyperuricemia and can precipitate a secondary form of gout.32
Disturbances in hemostasis also occur with polycythemia. Coagulation factors are commonly abnormal in patients with hematocrits in excess of 60%.33 Actual platelet counts may be normal but can be increased initially in some patients, with subsequent decreases related to persistent and worsening desaturation. There is evidence of shortened platelet survival time in patients with cyanotic heart disease.34 Laboratory evaluation of coagulation status requires that correction be made for the diminished volume of plasma and for the volume of anticoagulant used in blood samples to avoid false results. Hematologic management of adults with cyanotic congenital heart disease requires special experience and knowledge.35
The major consequences of cyanosis can be avoided in many instances, although differences in intelligence have been demonstrated between cyanotic and acyanotic children.36
Children with severe cardiac malformations frequently exhibit retardation of growth and development, with height and weight near or below the third percentile or weight 20 percentile points below the mean percentile for height.37
Growth retardation is most severe among children with overt cyanosis and those with large left-to-right shunts that cause heart failure. Heart failure tends to cause a greater reduction of weight than of height. Skeletal retardation, reflected by bone age, usually occurs with height and weight retardation and, among children with cyanotic heart disease, is correlated with the severity of hypoxemia.
Other factors contribute to growth retardation, including insufficient caloric intake, dyspnea, frequent infections, psychological disturbances, malabsorption, and hypermetabolism. Among infants with severe congenital heart disease recognized within the first year of life, there is a significantly increased incidence of subnormal birth weight, intrauterine growth retardation, and major extracardiac anomalies.3 Finally, a relatively small number of children have associated syndromes known to be characterized by growth retardation, such as rubella and Noonan, Turner, and Down syndromes. Growth retardation related primarily to congenital heart disease usually responds to surgical correction or palliation, with an impressive acceleration of growth and a return toward normal.
Although cardiac surgery is seldom recommended on the basis of growth failure alone, decelerated growth should be recognized early and, until proved otherwise, considered an index of the severity of heart disease. In general, the more successful the surgery, the less will be the retardation of growth and development, with its sequelae of physical, psychological, and intellectual problem.38
Pulmonary arterial hypertension (PAH) and pulmonary vascular obstructive disease (PVOD) are serious complications of congenital heart disease. PAH usually results from direct transmission of systemic arterial pressure to the right ventricle or pulmonary arteries via a large communication. Less frequently, it is caused by severe obstruction to blood flow through the left side of the heart at the pulmonary venous level or beyond. PVOD refers to a process involving structural and developmental changes in the smaller muscular arteries and arterioles of the lung that gradually diminishes and eventually destroys the ability of the pulmonary vascular bed to transport blood from the larger pulmonary arteries to the pulmonary veins without an abnormal elevation of proximal pulmonary arterial pressure.
Pulmonary resistance (Rp) may be as high as 8 to 10 Wood units immediately after birth, but falls rapidly throughout the first week of life. Indexed Wood units, as a measure of resistance to flow across either the pulmonary or the systemic vascular bed, are obtained by dividing the mean pressure difference (in millimeters of mercury) across the pulmonary or systemic vascular beds by the blood flow index (expressed in liters per minute per square meter) across those respective beds. By 6 to 8 weeks, it usually has reached the normal adult level (1-3 Wood units). These changes are accompanied by a gradual dilatation of the smaller followed by the larger muscular pulmonary arteries. In the weeks and months that follow, a thinning of the muscular walls occurs, with the growth of existing arteries and the development of new arteries and arterioles. The latter process contributes more than 90% of the smaller or intraacinar pulmonary arterial vessels present in older children and adults.39
Increased pulmonary arterial pressure has an adverse effect on the normal maturation of the pulmonary vascular bed. Such pressure promotes persistence of the thick muscular medial layer present in the smaller pulmonary arteries of term newborns, stimulates an extension of smooth muscle into smaller and more peripheral arteries than normal for age, and retards the growth of existing acinar arteries and the development of new vessels.
In the presence of a large systemic-to-pulmonary communication, pulmonary arterial pressures remain at or near systemic levels, with the result that the diminution in pulmonary muscle mass and pulmonary resistance is less rapid and of a lesser magnitude than in a normal infant. Nevertheless, the reduction in resistance is usually sufficient to permit a large pulmonary blood flow with resulting heart failure by the end of the first month. Exceptions are found among infants with a large systemic-to-pulmonary communication living at high altitude with alveolar hypoxia—a stimulus for pulmonary vasoconstriction—in whom there is less than normal involution of the medial musculature and decrease in pulmonary vascular resistance. Clinically, these infants may lack signs of heart failure. It is also seen in some children with Down syndrome and a large VSD or AV canal who may hypoventilate or have upper airway obstruction. Rarely, an infant will maintain a very high pulmonary vascular resistance in the face of an anatomically large systemic-to-pulmonary communication without evidence of significant hypoxemia or acidemia and remain free of the signs and symptoms of congestive failure. Conversely, in a premature infant in whom the medial muscle mass is less at birth than found in a full-term infant, the fall in pulmonary vascular resistance is usually much more rapid than normal.
Chronic PAH, increased flow, or both produce a characteristic series of histologic changes in the smaller pulmonary arteries and arterioles, originally described and graded by Heath and Edwards40 and by Rabinovitch,39 extending from medial hypertrophy present at birth to progressive obstruction and terminating with necrotizing arteritis within the media. Rabinovitch further refined the pathologic characterization based on the degree of extension into peripheral vessels.
Estimation of pulmonary vascular resistance from data obtained at cardiac catheterization remains the most widely used means of assessing the state of the pulmonary vascular bed. Hypoxemia from oversedation, atelectasis, or pneumonitis at the time of study may confound interpretation of the hemodynamic data. If pulmonary vascular resistance is elevated, responsiveness to vasodilation induced by the inhalation of 100% oxygen, the pulmonary arterial administration of prostacyclin, or the inhalation of nitric oxide should be tested.41
Values of Rp ≥3 Wood units are considered normal. The status of the pulmonary vasculature also can be expressed as a ratio of pulmonary vascular resistance to systemic vascular resistance (Rp/Rs). Pulmonary/systemic resistance ratios less than 0.2:1 are considered normal.
As pulmonary vascular resistance increases, pulmonary blood flow generally decreases. Eventually, a point is reached where surgical closure of the defect will produce only a small decrease of blood flow, a proportionately small decrease in pulmonary arterial pressure, and no significant change in the factors contributing to the progression of vascular disease. At this point surgery usually is not recommended, as the benefits are minimal and closure of the defect may eliminate a useful “blow-off” for increasing resistance. An Rp/Rs ratio of 0.7:1 or an Rp of 11 Wood units with a pulmonary-to-systemic blood flow ratio of 1.5:1 is the criterion generally used to define this situation. Without surgery, these patients survive as examples of the Eisenmenger syndrome, in which Rp ≥ Rs and at least some right-to-left shunting occurs at rest or with exercise. Some of these patients can survive for several decades and lead productive lives, with relatively mild symptoms and few limitations.42
The decision regarding surgery for patients with less severe PVOD is a clinical one. The higher the calculated resistance, the greater the structural changes in the pulmonary vasculature (as judged by lung biopsy or quantitative pulmonary arterial wedge angiography), and the older the patient with any given level of elevated resistance or grade of structural change, the less likely it is that the outcome of surgery will be satisfactory.39
The prevention of PVOD requires the identification of the patients at risk—that is, all patients with a systemic-to-pulmonary communication and a pulmonary arterial systolic pressure higher than half the systemic arterial systolic pressure. Also included are all patients with transposition, regardless of pressure or flow, with the possible exception of those with severe pulmonary stenosis. Ideally, all patients at risk should undergo correction of the lesion or protection of the pulmonary arterial bed within the first year of life unless there is proof that the pulmonary arterial systolic pressure has fallen to or is less than half the systemic systolic pressure among those with normally related great arteries. Among patients with transposition with a large VSD or patent ductus arteriosus, action must be taken within the first 3 months of life.
With advances in the surgical treatment of congenital heart defects, more patients are living to adulthood. This discussion of potential long-term problems is intended for those who follow these children after surgery and through adult life43 (see Chap. 84). Residua, sequelae, and complications result from most surgical procedures for congenital heart defects. A residual part of the original defect, such as mitral prolapse in repaired ASD, may purposefully not have been approached surgically. Some sequelae are unavoidable consequences of the intervention, such as pulmonary regurgitation after balloon dilation valvuloplasty or surgical valvotomy. There are also complications that occur as unexpected but related events after successful surgery, such as late complete heart block. When viewed with these possibilities in mind, only surgical ligation or coil occlusion of a PDA is likely to result in no long-term problems.
Most patients have residual murmurs after surgery for congenital heart defects. Determination of the origin of these murmurs and evaluation of the severity of the hemodynamic abnormalities they represent are important. Noninvasive diagnostic tools, especially two-dimensional echocardiography and Doppler, are often useful. The risk of infective endocarditis persists for 6 months in all patients after surgery. Patients with residual VSD patch leaks, prosthetic valves, and conduits have increased risk of endocarditis.44
There are specific problems related to some of the more common defects. For those with repaired ASDs, VSDs, and AV (canal) septal defects, a residual shunt may be present, but ordinarily it is small and not hemodynamically significant. Those with repaired AV canal defects may have important AV valve regurgitation. Repaired coarctation of the aorta can gradually become narrowed again, or patients may develop systemic hypertension. Intervention for valvular pulmonary stenosis usually results in mild residual stenosis and regurgitation, which are well tolerated and have little tendency to progress with time. The natural history of valvular aortic stenosis after intervention is not as benign.45,46
Significant regurgitation must be avoided, so the initial results may not be as good in terms of the severity of residual stenosis. In addition, because aortic stenosis tends to worsen with time, proper follow-up is mandatory for these patients.
Few patients enter adulthood with the continued problem of cyanosis, as those with defects amenable to surgical correction should have had surgery well before this time. Only patients with complex and irreparable defects and those with pulmonary vascular disease should experience problems of cyanosis during the adult years. Particularly important among these patients is management of any attendant psychosocial problems (employment, insurability,47 and learning disabilities) and difficulties related to pregnancy.48 Those who have had surgery for cyanotic defects are more likely to have sequelae and complications. Some degree of exercise intolerance is not unusual in this group of patients, and exercise stress testing aids in their management.49
Dysrhythmias are particularly common among these patients. In patients who have had intraventricular repairs, most commonly for tetralogy of Fallot, late complete heart block and serious ventricular arrhythmias can occur and may result in sudden death.50 This risk appears to be highest in those who had transient complete heart block at the time of surgery and who develop right bundle-branch block with left anterior hemiblock after surgery. Extensive intraatrial surgical procedures for transposition of the great arteries also frequently lead to dysrhythmias, most commonly sick sinus syndrome with brady- tachyarrhythmias and atrial flutter, with a high incidence of sudden death.51 Ambulatory 24-hour ECG monitoring, stress testing (see Chap. 16, and intracardiac electrophysiologic studies52 are important in following patients who have had complex repairs. Atrial enlargement after the Fontan operation has resulted in atrial flutter and/or fibrillation, which are frequently therapeutically challenging.53
Serious ventricular dysfunction54 and venous obstruction also may occur. Despite intraatrial baffle repairs for transposition of the great arteries, the anatomic right ventricle must perform systemic work, leading to potential compromise of ventricular performance.55 In addition, these repairs may lead to pulmonary and/or systemic venous obstruction. Atriopulmonary and more recently cavopulmonary connections for the repair of tricuspid atresia and many types of univentricular hearts frequently leave an anatomically abnormal ventricle as the systemic ventricle. In this group of patients, the absence of a ventricle on the pulmonary side of the circulation results in elevated systemic venous pressure, systemic venous hypertension, and possible protein-losing enteropathy.56
Finally, some children have had repairs with synthetic prostheses. Because artificial valves cannot keep pace with somatic growth of the child, long-term stability is critical. Conduits with or without valves placed at surgery can degenerate or become obstructive. Bioprosthetic valves undergo accelerated fibrosis and calcification in patients younger than approximately 30 to 35 years of age. In spite of these problems, the majority of patients who reach adulthood after surgical repair of congenital defects are relatively asymptomatic; they can and do lead productive lives.
Intracardiac Communications between the Systemic and Pulmonary Circulations, Usually Without Cyanosis
A VSD is the most common congenital cardiac anomaly. It may be an isolated defect or part of a complex malformation. Approximately 80% of these defects are perimembranous but may extend into the inlet, trabecular, or outlet sections of the muscular septum. Less common are conal septal or subarterial doubly committed defects (5%-7%), inlet defects lying beneath the septal leaflet of the tricuspid valve in the region of the atrioventricular canal (5%-8%), and defects in the muscular septum that may be in the inlet, trabecular, or outlet area (Fig. 83–4).57 Multiple muscular defects are not infrequently seen.
Figure 83–4.

Different types of ventricular septal defects when viewed from the right ventricle. Atrioventricular (AV) canal defects occur immediately adjacent to the tricuspid and mitral valves in the portion of the ventricular septum derived from the embryonic endocardial cushions. Membranous defects occur more anteriorly in the portion of the ventricular septum adjacent to the tricuspid and aortic valves that in the normal heart is composed of a membrane. Subpulmonary and other outflow-type defects occur due to abnormal alignment or muscular defects in the conus. Reproduced with permission from Fyler DC, ed. Nadas’ Pediatric Cardiology. Philadelphia, PA: Hanley & Belfus; 1992.
The incidence of VSDs is approximately 2 per 1000 live births, and its prevalence among school-age children has been estimated as 1 per 1000, constituting approximately one-quarter of the congenital cardiac malformations in combined series (see Table 83–1). Males and females are affected equally.
VSDs may be isolated or associated with other congenital cardiac abnormalities. Malformations associated with VSD are, in order of decreasing frequency, (1) coarctation of the aorta; (2) additional shunts, most commonly ASD and PDA; (3) intracardiac obstructions such as subpulmonary or subaortic stenosis, mitral stenosis, and anomalous muscle bundle of the right ventricle; and (4) incompetent atrioventricular valves.
The consequences of a VSD depend on the size of the defect and the pulmonary vascular resistance. A small defect offers a large resistance to left-to-right shunting. There is no elevation of right ventricular or pulmonary arterial pressure, and the defect may be so small that it can be detected only by two-dimensional imaging with Doppler color-flow mapping or selective left ventricular angiography. This type of defect imposes no physiologic cardiac burden, although the risk for infective endocarditis exists, but is very small. Moderate-sized defects result in a difference between the right and left ventricular systolic pressures but may allow a large left-to-right shunt, producing left atrial hypertension and/or dilation and left ventricular volume overload. The development of pulmonary vascular disease among these patients is unusual but possible.
When the effective area of the defect is large, there is virtually no resistance to flow, and systemic systolic pressure is present in the right ventricle and the pulmonary artery. The relative resistance of the two vascular beds governs the proportion of blood entering the two circulations. At birth, pulmonary vascular resistance is high, with little if any left-to-right shunt, despite the presence of a large defect. This resistance gradually falls over the first few weeks of life, permitting a progressively greater amount of blood to flow through the defect, to the pulmonary arteries and back to the left atrium and left ventricle. In many infants, the left ventricular volume overload eventually leads to left ventricular “failure,” with elevated left ventricular end-diastolic and left atrial pressures and pulmonary congestion.
Infants or children with a small isolated defect are asymptomatic. The murmur of a small defect may be detected within the first 24 to 36 hours of life, as the very restrictive opening permits the normal rapid fall in pulmonary arterial resistance and pressures. In term infants born at sea level with a large VSD, clinical deterioration may occur at any time from approximately 3 to 12 weeks after birth. In premature infants, pulmonary vascular muscular hypertrophy regresses more rapidly, with failure frequently noted at 1 to 4 weeks. Parents describe tachypnea, grunting respirations, and fatigue, particularly with feedings. Weight gain is slow, and excessive sweating is common.
A child with a small defect is comfortable. If the defect is small, the pulmonary artery pressure is normal, so the second heart sound is not accentuated. The systolic murmur along the lower left sternal border is characteristically holosystolic but may be limited to early or midsystole. This latter feature suggests a defect in the muscular septum rather than the membranous septum. With moderate-sized communications, a systolic thrill at the lower left sternal border is common.
Infants with large defects, large left-to-right shunt, and PAH tend to be restless, irritable, and underweight. Moderate respiratory distress may be present. Both the right and the left ventricular systolic impulses are hyperdynamic to palpation. A thrill at the lower left sternal border is common. The second heart sound is narrowly split, with a loud, frequently palpable pulmonary component. Third heart sound gallops at the apex are common. Characteristically, the murmur is holosystolic at the lower left sternal border and is accompanied by a mid-diastolic rumble of grade 2 to 3 intensity at the apex, with the latter indicating a pulmonary-to-systemic blood flow ratio (Qp/Qs ratio) of 2:1 or greater. Hepatic enlargement can be identified below the right costal margin. Pulmonary rales may be heard with severe failure.
Over several months, the shunt may diminish with improved rate of weight gain, less tachypnea, diminution of the precordial hyperactivity, and disappearance of the apical diastolic flow rumble. This clinical improvement may be a result of the defect becoming smaller, the development of subvalvular pulmonary stenosis with little or no appreciable change in the size of the defect, or, most worrisome, the development of PVOD with continued severe PAH. With developing subpulmonary stenosis, the systolic murmur radiates toward the upper left sternal border and the second heart sound becomes more widely split, with a progressive diminution in the intensity of the pulmonary component. Decreased shunting resulting from pulmonary vascular disease is characterized by a gradual reduction in the intensity and duration of the systolic murmur, narrow splitting of the second heart sound, and marked accentuation of the pulmonary component.
The clinical picture of advanced pulmonary vascular disease from a congenital left-to-right shunt (Eisenmenger syndrome) is that of a relatively comfortable older child, adolescent, or young adult with mild cyanosis and clubbing with a prominent a wave in the jugular venous pulse, a right ventricular parasternal heave, and a narrow split or single second heart sound with an accentuated pulmonary component. An early pulmonary systolic ejection sound reflecting dilatation of the main pulmonary artery may be present, and there may be no systolic murmur at all. In older adolescents and adults, an early diastolic murmur of pulmonary regurgitation or a holosystolic murmur of tricuspid regurgitation may appear.
In the presence of a small defect, the heart size and the pulmonary blood flow are barely altered. With large defects, there is moderate to marked enlargement of the heart, with prominence of the main pulmonary arterial segment and overcirculation in the peripheral lung fields. The left atrium is dilated unless an associated ASD is present, allowing decompression of the left atrium. With increasing pulmonary vascular disease, there is regression of the heart size toward normal, whereas the central pulmonary arteries remain dilated. The peripheral pulmonary arterial markings become attenuated, and a “pruned” effect is produced in the outer third of the lung fields.
With a small defect, there is normal progression of the mean QRS axis from right to left and the gradual decrease of the prominent right ventricular voltages characteristic of newborns. The left ventricular forces remain within normal limits or become slightly augmented as a reflection of the mild left ventricular volume overload. With large defects, the mean QRS axis tends to remain oriented to the right, with little or no regression in right ventricular voltage. The left ventricular forces gradually increase, resulting in a pattern of biventricular hypertrophy within the first few weeks of life. Left atrial enlargement is usually present, as is right atrial enlargement. With the development of either pulmonary vascular disease or progressive pulmonary stenosis, the mean QRS axis tends to remain oriented to the right; there is no regression in right ventricular voltage, but left ventricular and left atrial hypertrophy lessens or disappears.
Two-dimensional imaging can distinguish an uncomplicated VSD from more complex malformations and is capable of imaging most defects directly when multiple transducer positions are used. The addition of pulsed-wave Doppler with color-flow mapping permits the identification of small, multiple, muscular, and other less easily visualized defects. The position and size of the VSD can be determined, as well as its relationships to the aorta, pulmonary artery, and AV valves. Continuous-wave Doppler interrogation of flow across the defect can estimate the systolic right ventricular pressure as the difference between the systolic pressure measured by a blood pressure cuff (in the absence of aortic stenosis) and the Doppler gradient (Fig. 83–5). In the absence of pulmonic stenosis, the right ventricular systolic pressure provides an estimate of the pulmonary artery pressure. Right ventricular systolic pressure also can be estimated by measuring the right ventricular to right atrial systolic pressure gradient across the tricuspid valve in the presence of tricuspid regurgitation.58
Figure 83–5.
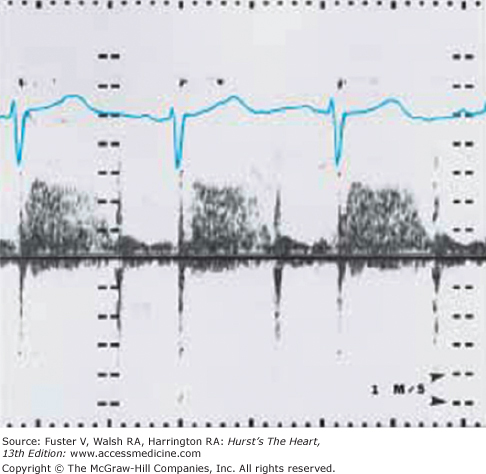
Continuous-wave Doppler with spectral display from the left lower sternal border of a child with a ventricular septal defect that demonstrates holosystolic turbulence with peak velocity = 2.8 m/s across the defect, compatible with an instantaneous systolic pressure difference of 31 mm Hg between the right and left ventricles.
Though cardiac catheterization is performed infrequently in infants with isolated VSDs, these studies show an increase in oxygen saturation at the right ventricular level and pulmonary artery level, reflecting the left-to-right ventricular shunt. With small defects, the right ventricular and pulmonary arterial systolic pressures are normal. With large defects, these pressures are at or near systemic levels, and the mean left atrial pressure may be elevated to the range of 10 to 15 mm Hg.
Selective left ventricular angiography in the anteroposterior, lateral, and oblique views with craniocaudal angulation is used to establish the spatial relations of the great arteries to each other and to the ventricles and also to determine the exact site, size, and number of septal defects (Fig. 83–6). Aortography is helpful in eliminating the possibility of an associated ductus arteriosus or unsuspected coarctation of the aorta if the arch cannot be imaged adequately by echocardiography.
Figure 83–6.
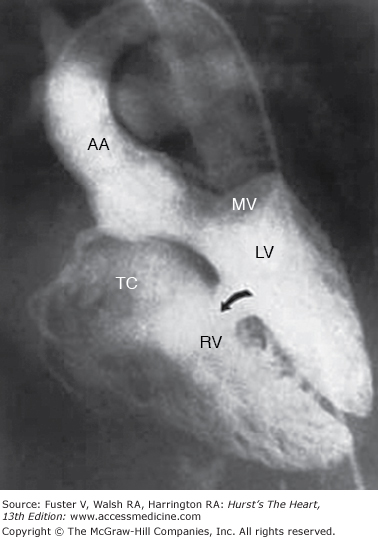
Multiple trabecular ventricular septal defects (VSDs). Retrograde left ventriculogram, four-chamber projection, profiles the mitral and tricuspid valves, and the midtrabecular VSD (arrow). Additional VSDs closer to the apex are more anterior in location and are not profiled in this projection. AA, ascending aorta; LV, left ventricle; MV, mitral valve; RV, right ventricle; TV, tricuspid valve. Reproduced with permission from Lock JE, Keane JF, Perry SB. Diagnostic and Interventional Catheterization in Congenital Heart Disease. 2nd ed. Boston, MA: Kluwer; 2000.
The majority of VSDs are small and do not present a serious clinical problem. Approximately 25% of these small defects close spontaneously by 18 months, 50% by 4 years, and 75% by 10 years.59 A spontaneous closure rate approaching 45% within the first 12 to 14 months has been observed among infants with an uncomplicated perimembranous or muscular VSD in the neonatal period.60 Even large defects tend to become smaller, but the likelihood of eventual spontaneous closure is much lower (probably in the range of 60%) if defects are large at 3 months of age and only 50% percent if it is still large at 6 months.61
Heart failure is an almost inevitable complication of a large VSD. Approximately 80% of infants with large defects require hospitalization by age 4 months.3 Significant subvalvular pulmonary stenosis develops in approximately 3% of these individuals and if progressive may eventually result in right-to-left shunting at the ventricular level. Associated pulmonary arterial hypertension is generally reversible in the first 12 months of life; thereafter, it becomes less likely to regress. Infants and children with a pulmonary systolic pressure in excess of 50% of the systemic arterial systolic pressure beyond the first year of life are at risk for this complication.61 A very small number of infants with large VSDs maintain a high level of pulmonary vascular resistance throughout the first year of life and remain almost entirely free of symptoms of heart failure. In these patients, irreversible pulmonary vascular disease may develop without the usual and expected clinical signs and symptoms described above.
A small number of children, 0.6% in a large group of carefully followed patients, develop aortic regurgitation as a result of prolapse of either the right or posterior aortic valve leaflets into the defect.62 This complication is more prevalent among males than females, in a ratio of 2:1, and seems particularly likely to occur with defects of the subpulmonary type, but is also seen with perimembranous VSDs. Shunt size appears not to be related to the development of this complication. The characteristic aortic diastolic murmur may appear at any time between ages 6 months and 20 years. Regurgitation is usually progressive, sometimes rapidly so.
The basis of the medical management of children with VSDs is an understanding that defects frequently narrow and may close spontaneously. Approximately 70% of small VSDs probably close.59 Even large muscular defects may become smaller, and up to 25% of them will become hemodynamically insignificant if one can wait long enough.
For children with a large VSD, the first decision point usually occurs before 8 to 12 weeks of age. Infants with large septal defects generally develop significant left-to-right shunts as the pulmonary resistance drops. Heart failure ensues with tachypnea, tachycardia, and feeding difficulty. Digoxin and diuretics are occasionally useful, but if the left-to-right shunt is very large, feeding may remain problematic. For children who cannot gain weight of at least 15 g/d (30 g/d is normal) without other cause for failure to thrive, surgical repair is indicated. Increasing the caloric density of the formula from 20 cal/oz to as much as 32 cal/oz to provide at least 150 kcal/kg daily may be successful. Children unable to take more than 10 to 12 oz/d, however, will most likely require surgical repair.
Children with unrestrictive or mildly restrictive VSDs have pulmonary artery hypertension that may lead to irreversible pulmonary vascular obstructive disease. Although some of these children will gain adequate weight, if the pulmonary artery pressure remains elevated at 9 to 12 months of age, surgery is indicated. Signs suggestive of normal or mildly elevated pulmonary artery include a normal pulmonary component of the second heart sound, the absence of right ventricular hypertrophy on ECG, and a large intraventricular pressure gradient by echocardiography. Infrequently, for children for whom signs, symptoms, and laboratory findings are ambiguous or conflicting, cardiac catheterization may be necessary to assure that the pulmonary artery pressure is normal and that pulmonary vascular obstructive disease is not a risk.
A final decision point occurs somewhere between 5 to 10 years of age. Although the defect has not caused failure to thrive or pulmonary hypertension, it may still produce a significant left-to-right shunt and left ventricular volume overload. There is no firm criteria that suggest a dangerous level of left ventricular volume overload, but many centers proceed with VSD closure when the pulmonary-to-systemic flow ratio (measured by cardiac catheterization, radionuclide angiography, echocardiography, or magnetic resonance imaging) is more than 2:1. Others use significant left atrial and left ventricular dilation by echo. A minority of centers do not recommend surgical closure as long as the pulmonary artery pressure is normal because there are few adults with a VSD who develop late congestive heart failure.
Unfortunately, not all patients with a large defect are identified early in life when it is possible to prevent injury to the pulmonary vascular bed. If significant PAH persists, progression to irreversible pulmonary obstructive disease can ensue. For this reason, with late presentation, prompt surgical closure of defects is recommended in all individuals older than the age of 2 years if the pulmonary arterial systolic pressure is greater than half the systemic arterial systolic pressure, the mean pulmonary pressure exceeds 25 mm Hg, or the Rp/Rs ratio is higher than 0.3:1. With severe pulmonary vascular disease, a point eventually is reached at which the risk of death at operation or in the months or years immediately after the operation as a result of progressive vascular disease more than offsets the possible benefits from surgical closure. At present, surgery is recommended if the calculated Rp is less than 10 Wood units/m2 or the Rp/Rs ratio is 0.7:1, provided that the Qp/Qs ratio is at least 1.5:1. In adults, the upper limit of pulmonary vascular resistance for surgery is approximately 10 Wood units.
Patients in whom the defect is judged clinically to be small at 6 months of age may be reexamined at 1- or 2-year intervals; however, the change in subacute bacterial endocarditis recommendations makes the need for long-term follow-up of questionable benefit in those with muscular VSDs. For perimembranous defects or subpulmonary defects, continued follow-up is indicated to assess for the development of subaortic stenosis or aortic valve prolapsed with regurgitation.
In patients with Eisenmenger complex,42 stamina is limited by systemic arterial hypoxemia and, in some, right-sided heart failure. Complications to be anticipated include syncope, hemoptysis, brain abscess, hyperuricemia, and heart failure. Pregnancy, with a maternal mortality of 30% to 60%, and oral contraceptives are contraindicated. Transient symptomatic relief from extreme polycythemia (usually >65%) may be achieved with careful erythropheresis. Travel to or living at high altitudes is poorly tolerated, and supplemental oxygen should be provided and used during air travel. A recent study reports that the average age of death for individuals with Eisenmenger complex related to VSD is 43 years, with heart failure the cause of death in the majority.63
The risk of congenital heart disease for a subsequent sibling of a single affected child is on the order of 1% to 2%. The risk to a newborn of a parent with VSD increases to 3%.64
Previously, banding of the pulmonary artery to reduce pulmonary blood flow and pressures played an important role in the management of congestive heart failure and the prevention of PVOD. Because surgical closure of VSDs in infancy is nearly uniformly successful, banding is used rarely. Complications of pulmonary arterial banding include deformity of the pulmonary arteries and/or pulmonary valve, progressive right ventricular hypertrophy with loss of ventricular compliance, and the development of subaortic left ventricular outflow tract obstruction.
Surgical closure of VSDs in infancy is nearly uniformly successful using cardiopulmonary bypass with cardioplegic arrest and moderate systemic hypothermia. Total circulatory arrest or minimal perfusion with profound hypothermia is sometimes necessary in infants who weigh less than 5 kg.65,66 Perimembranous VSDs may be exposed through the right atrium and the tricuspid valve orifice. A transverse or longitudinal right ventriculotomy may be necessary for closure of high conal septal defects associated with aortic valve leaflet prolapse. Care is required to prevent injury to the AV node near the ostium of the coronary sinus and to the bundle of His as it courses inferiorly, passing on the left side of the ventricular septum near the posterocaudal margin of the septal defect. Intraoperative transesophageal echocardiography with Doppler color-flow assessment can be used for the detection of significant residual shunting or previously unsuspected problems that may be corrected in the operating room.
Results from primary surgical closure of VSDs are generally excellent. Surgical mortality is less than 1% in centers with extensive experience, when surgery is performed during the early months of life before the evolution of PVOD. Operative risk should be even lower in older children if the pulmonary vascular resistance remains low. The pulmonary vascular bed responds favorably when the systemic-to-pulmonary shunt is eliminated before age 2 years. Normal life expectancy and functional capabilities should be anticipated postoperatively. Survival 25 years after the closure of a VSD is approximately 95%.66 The mortality rate is unquestionably higher among patients who are operated on with Rp >7 Wood units. In such cases, surgeons have employed fenestrated patch closure, which retains a restrictive ventricular communication to provide a “pop-off” right-to-left shunt postoperatively. In the setting of suprasystemic pulmonary artery pressure, the physiologic intent is to maintain systemic cardiac output while reducing the potential for right ventricular failure.
The surgical repair of a multiple muscular VSD has been more problematic. The highly trabecular right ventricular septal surface can make the localization of all the defects difficult. A new approach has achieved successful closure from a right ventricular apical infundibulotomy.67,68 Device closure of these defects, as well as perimembranous communications, are now possible either in the catheterization laboratory.69-74 The early results of transcatheter closure of perimembranous and muscular VSDs have been very satisfactory, with an overall closure rate of 95.3% among multiple centers as reported by the European Registry. The most frequent complication encountered was development of complete AV block in 3.7%, most of which occurred in perimembranous VSDs. Device embolization, infection, and vascular complications were infrequent. A single death was reported among the cohort of 430 patients.
In a novel approach, closure of muscular septal defects using devices has been accomplished during surgical repair in so-called hybrid procedures, both for isolated muscular VSDs and for those found in combination with other complex lesions.75
An atrial septal defect is a communication between the atria resulting from a deficiency of tissue in the septum, as distinguished from a patent foramen ovale, where septum primum, though preserved, is not adherent to the superior limbic band of septum secundum.
ASDs are found in various sizes, some sufficiently large to allow free communication between the atria. They may be subdivided according to anatomic location (Fig. 83–7).76
Figure 83–7.
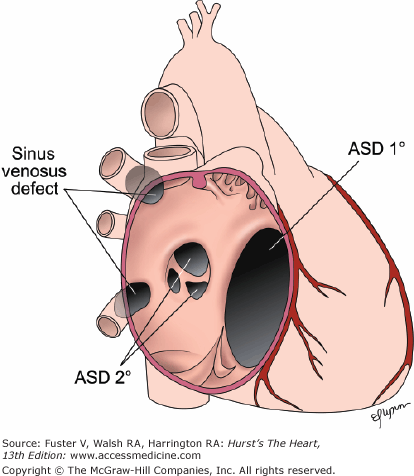
Types of interatrial communications. Secundum type atrial septal defects (ASD 2°) are located near the fossa ovalis in the center of the atrial septum. An ostium primum type atrial septal defect (ASD 1°) is located immediately adjacent to the tricuspid and mitral valves, a portion of the atrioventricular canal. Sinus venosus defects, considered “unroofing” defects between the right pulmonary veins and the right atrium, are located in the area derived from the embryologic sinus venosus. IVC, inferior vena cava; RPVs, right pulmonary veins; SVC, superior vena cava. Reproduced with permission from Keane JF, Geva T, Fyler DC. Atrial septal defect. In: Keane et al.5
This defect classically involves the region of the fossa ovalis and is the most common type (70%; see Fig. 83–7).76,77 Atrial septal tissue separates the inferior edge of the defect from the AV valves. Associated partial anomalous pulmonary venous connections are not uncommon, with one or more of the right pulmonary veins draining into the right atrium or one of its tributaries.
Defects of the AV septum lying inferior to the fossa ovalis constitute approximately 20% of ASDs and are part of a complex malformation known as common atrioventricular canal defects, which are considered below (see Fig. 83–7).
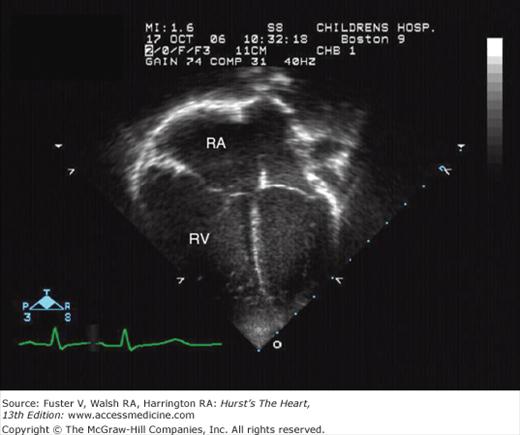
These defects, accounting for approximately 6% of the total, appear to represent a biatrial connection of the superior vena cava (or, in rare instances, the inferior vena cava), which straddles the otherwise normal intact atrial septum. In some cases, one or more of the right-sided pulmonary veins course anomalously draining to the vena cava or directly to the right atrium near its junction with the vena cava (see Fig. 83–7).
A coronary sinus defect is an uncommon type of ASD located in the position normally occupied by the ostium of the coronary sinus. This defect is part of a developmental complex consisting of the absence of the coronary sinus and entry of the left superior vena cava directly into the left atrium.
With sizable shunts, the right atrial and ventricular chambers, as well as the central pulmonary arteries, become enlarged (Fig. 83–8). Pulmonary hypertension may develop, but not before the third decade. The earliest lesion of pulmonary hypertensive change is cellular fibrous with intimal thickening in the proximal segments of arterioles. The pulmonary arterial pressure then rises, followed by the development of medial hypertrophy of muscular arteries and the appearance of plexiform lesions. The right ventricular wall hypertrophies, and atherosclerosis may occur in the major pulmonary arteries. Saccular aneurysm and thrombosis with dissecting aneurysm or rupture may occur (see Pulmonary Arterial Hypertension and Pulmonary Vascular Obstructive Disease above). In the final state, the pulmonary vascular bed may be difficult to distinguish from that in VSD with PVOD.
With moderate to large defects, no resistance to blood flow across the defect is present, resulting in equalization of pressure between the 2 atria. A left-to-right shunt of blood occurs (see Fig. 83–8) because (1) the right atrium is more distensible than the left, (2) the tricuspid valve is normally more capacious than the mitral valve, and (3) the thin-walled, compliant right ventricular chamber accommodates a larger volume of blood at the same filling pressure than does the left ventricle. A large left-to-right shunt is found only after the right ventricular compliance has had time to change appreciably from that of the left ventricle. Presumably, a fall in pulmonary vascular resistance leads to an increase in right ventricular stroke volume, a decrease in end-systolic volume, and enhanced right ventricular diastolic filling.78 Most patients tolerate a large volume load on the right ventricle and pulmonary circuit for many years. With the development of pulmonary vascular disease and PAH, the left-to-right shunt decreases, largely because of the increased thickness and decreased compliance of the right ventricle. In some patients, this process continues until the shunt reverses, with resulting arterial desaturation and cyanosis.
ASD is found in approximately 6% of children with congenital heart disease who survive beyond the first year of life.5 After bicuspid aortic valve, it is the most common form of congenital heart disease among adults.
ASDs have a female-to-male ratio of approximately 2:1. The mode of transmission is best explained in most instances on a multifactorial basis, in which the risk would be approximately 2.5% for first-degree relatives of a single affected family member. However, examples of autosomal-dominant transmission are recognized,79 either as an isolated entity associated with severe AV conduction disturbances or with upper extremity malformations, as in the Holt-Oram syndrome.
The majority of these children are considered asymptomatic but probably most have some mild diminution of stamina. Often the patient or the parents comment on the increased endurance that follows surgical correction. Symptoms of mild fatigue and dyspnea tend to be recognized in the late teens and early twenties, and at least three-quarters of these individuals will be symptomatic as adults. Heart failure is rare in childhood, becoming more prevalent in the fourth and fifth decades of life, and may be associated with the onset of arrhythmias.80
Normal growth and development are the rule. In the older child, prominence of the left anterior chest is common, and a hyperdynamic right ventricular systolic lift can be felt. The jugular venous pulse demonstrates that the v wave is equal to the a wave rather than the normal a wave predominance. The first heart sound may be slightly accentuated at the lower left sternal border. The two components of the second heart sound are characteristically widely split and fixed, without respiratory variation. The pulmonary component of the second heart sound may be accentuated, more so in the presence of PAH. With increasing pulmonary arterial pressure and resistance, the interval between the aortic and pulmonary components of the second heart sound narrows and the pulmonary component becomes louder, but the lack of respiratory influence on the interval between the two components persists. A midsystolic crescendo–decrescendo murmur of grade 2 to 3 intensity is heard at the left upper sternal border, reflecting increased right ventricular stroke volume and relative pulmonary stenosis. A low- to medium-pitched early diastolic murmur over the lower left sternal border, denoting increased diastolic flow across the tricuspid valve, is present in most individuals with large shunts (see Chap. 16). Cyanosis and clubbing reflect right-to-left shunting. In this setting, the murmurs of tricuspid and pulmonary regurgitation are not uncommon.
Mild to moderate cardiac enlargement and prominence of the main and branch pulmonary arteries are characteristic (Fig. 83–9).
An rsR′ pattern over the right precordial leads indicating right ventricular conduction delay or mild right ventricular hypertrophy is characteristic though not pathognomonic of secundum-type ASD. A qR pattern in this lead is indicative of right ventricular hypertrophy related to volume overload. The mean QRS axis in the frontal plane is 90 degrees or greater in 60% of patients. Left-axis deviation is common in primum-type ASD. Abnormal leftward p axis is often present in sinus venosus-type ASD. Atrial fibrillation and atrial flutter are usually limited to adults.
Two-dimensional and Doppler echocardiography with color-flow mapping (see Chap. 18) permits identification and visualization of secundum, AV canal, and sinus venosus defects. Visualization of anomalous draining pulmonary veins is slightly more difficult. The transesophageal approach offers excellent images for those patients in whom the transthoracic approach is inadequate.81 Three-dimensional echocardiograms have been shown to provide excellent images of the atrial defects82,83 (Fig. 83–10).
Figure 83–10.

Three-dimensional echocardiogram of a secundum atrial septal defect (ASD; arrow). This is a right atrial en-face view that shows the size, shape, and position of the defect in relation to the right atrial septal surface. SVC, superior vena cava; IVC, inferior vena cava; TV, tricuspid valve.
There is an increase in oxygen saturation in the right atrium, right ventricle, and pulmonary artery compared with those obtained from the superior or inferior vena cava. Pulmonary arterial and right ventricular systolic pressures are normal or only slightly elevated. A systolic pressure gradient of up to 20 mm Hg across the right ventricular outflow tract is accepted as being secondary to flow rather than to anatomic obstruction. The right and left atrial mean and phasic pressures are virtually identical, with little if any elevation above normal (mean pressure gradient <3 mm Hg) unless there are associated abnormalities.
Defects of the secundum type are often undetected in the first year or two of life because of the lack of symptoms and the unimpressive auscultatory findings. A soft systolic murmur is the usual reason for referral. Symptoms become more common in persons in their late teens and twenties, and by age 40 years, the majority of these individuals are symptomatic, some severely so.84 Pulmonary vascular disease with serious pulmonary hypertension begins to make its appearance in persons in their early 20s. It affects approximately 15% of young adults, particularly women, and may be rapidly progressive, especially with pregnancy. The incidence of atrial fibrillation or flutter also increases with each decade and is closely linked to the onset of heart failure. Heart failure is the most common cause of death among patients without intervention. Other causes of death include pulmonary embolism or thrombosis, paradoxical emboli, brain abscess, and infection.
The few infants who present with symptoms of heart failure can be treated with anticongestive therapy. If the defect is uncomplicated and the symptoms persist, surgical closure is advised without further delay. For asymptomatic infants and children, closure is recommended at approximately 5 years of age. Restrictions of activity or exercise are unnecessary. If the physical, laboratory, and echocardiographic findings are completely characteristic, preoperative catheterization is unnecessary. Closure is recommended if the defect is associated with right ventricular volume overload by echocardiography. In those with pulmonary hypertension, closure is recommended for patients with Qp/Qs >1.5:1 by catheterization provided that the systemic arterial saturation is more than 92% and total Rp <15 Wood units.85 Closure is prudent before pregnancy or the use of contraceptives in view of the tendency to develop rapidly progressive PVOD in this setting. Transcatheter closure of centrally located secundum ASDs in older infants, children, and adults using various devices appears to be an acceptable alternative to surgical closure and is now the preferred method in many centers.86-89
Defects of the interatrial septum are exposed through the lateral wall of the right atrium. Ostium secundum (fossa ovalis) defects are frequently closed by direct suturing; very large defects or those with tenuous margins are closed with a patch, usually glutaraldehyde-treated autologous pericardium. Anomalous pulmonary veins are sought along the posterolateral aspect of the superior or inferior vena cava and from within the right atrium before closure of the defect. Sutures are placed with care along the posterior rim of the inferior vena caval orifice to prevent the creation of a tunnel from the inferior vena cava into the left atrium, which would cause postoperative hypoxemia.
High ASDs of the sinus venosus type, often associated with anomalous drainage of one or more right pulmonary veins into the superior vena cava (Fig. 83–11), are corrected by means of the placement of a pericardial or tubular Dacron patch from above the abnormally draining vein or veins down to and around the ASD. Pulmonary venous blood thus is diverted through the ASD into the left atrium. Pericardial gusset enlargement of the superior vena cava at the cavoatrial junction may be required. Anomalous right pulmonary veins draining to the right atrium are diverted into the left atrium by placement of a patch baffle well anterior and to the right of the pulmonary vein orifices. The risks of surgery are minimal (<0.5%), with short postoperative length of stay.
Figure 83–11.

Superior sinus venosus defect. High right parasternal border long-axis echocardiogram view in this patient with superior sinus venosus defect (SVD) demonstrates the absence of tissue (“unroofing”) defect between the right upper pulmonary vein (RUPV) and distal superior vena cava (SVC). Color Doppler flow demonstrates left-to-right flow from the RUPV to the SVC/RA, with mild flow acceleration across the defect. IVC, inferior vena cava; RA, right atrium.
In adults, clinical benefit after closure of ASDs can be anticipated even in those with physiologic compromise,90 but mortality is higher than it is in the young, and the magnitude of improvement is less certain. Nonetheless, closure of ASDs is advised even when Rp approaches 15 Wood units because of the excessive morbidity and mortality associated with a persistent interatrial communication.91
Although life-threatening complications after closure of ASDs in children are rare, transient postoperative atrial arrhythmias and postpericardiotomy syndrome with pericardial effusions occasionally are seen. The long-term prognosis for a normal life expectancy and functional capability is excellent for patients who have closure of an uncomplicated ASD during the first 2 decades of life.
Many centers are using devices to close atrial septal defects in the catheterization laboratory.86-89 This approach avoids potential surgical complications, reduces the length of hospitalization, and eliminates the midline sternotomy scar. Several devices are in various phases of investigation under US Food and Drug Administration protocol. Although not universally successful in closure of defects, device placement has achieved good results, particularly for those lesions with a sufficient circumferential rim of tissue. However, despite persistent shunting in ASDs with insufficient rims, late closure has approximated the early results found with ASDs having sufficient tissue. The uncertainty of long-term morbidity remains with regard to potential for rhythm disturbance and device erosion, so that longitudinal follow-up of each device is mandatory.
In partial anomalous pulmonary venous connection, one or more—but not all—of the pulmonary veins enter the right atrium or its venous tributaries. An ASD is usually present, but the atrial septum is rarely intact. Although there are many patterns of anomalous pulmonary venous connection, the four most common, in order of decreasing frequency, are (1) pulmonary veins from the right upper and/or middle lobe to the superior vena cava, usually with a sinus venosus ASD; (2) all the right pulmonary veins to the right atrium, usually in the polysplenia syndrome; (3) all the right pulmonary veins to the inferior vena cava, entering this systemic vein just above or below the diaphragm; and (4) the left upper or both left pulmonary veins to an anomalous vertical vein draining to the left brachiocephalic vein.
When the right pulmonary veins are connected to the inferior vena cava, the atrial septum may be intact. This venous anomaly may be isolated or may be part of the scimitar syndrome. That syndrome includes hypoplasia of the right lung, bronchial abnormalities, anomalous systemic pulmonary arterial supply to the right lung from branches of the descending thoracic and/or abdominal aorta, and dextroposition of the heart.
In an old autopsy series, partial anomalous pulmonary venous connection occurred in 0.6% of 801 cases,92 a much higher incidence than clinically suspected, suggesting that many cases may not be recognized during life. There is no sex predilection. Approximately 15% of all ASDs have this coexisting anomaly; however, in the case of the sinus venosus type, the association is in the range of 85%.
When partial anomalous pulmonary venous connection coexists with an ASD, the symptoms, as well as the other clinical manifestations, are indistinguishable from those of an isolated ASD. Isolated, uncomplicated anomalous connection of a single pulmonary vein usually goes undetected clinically, as in this circumstance only approximately 20% of the pulmonary venous flow returns to the right atrium or its tributaries. When the entire venous return from one lung or two pulmonary veins is connected anomalously, approximately 65% of the pulmonary venous flow returns to the right side of the heart, and the symptoms are similar to those of an ASD, with a comparable increase in pulmonary blood flow.
The findings are the same as those in patients with an ASD with the exception that the two components of the second heart sound, though usually widely split, move normally with respiration if the atrial septum is intact.
Right ventricular enlargement, pulmonary arterial dilatation, and increased pulmonary blood flow are characteristic when more than one pulmonary vein connects anomalously. With anomalous connection of the right pulmonary veins to the inferior vena cava, the pulmonary venous pattern may assume a crescent-shaped or scimitar curve in the right lower lung field along the right lower heart border (scimitar).
The ECG is normal (in the case of anomalous connection of a single pulmonary vein) or reflects volume overload of the right side of the heart, as was described above under Atrial Septal Defect.
If more than one pulmonary vein drains anomalously, the volume is usually sufficient to produce the characteristic pattern of right ventricular diastolic overload. Failure to visualize an atrial septal with two-dimensional imaging and color-flow mapping from a subcostal coronal or high right-sided parasternal longitudinal view should arouse suspicion of anomalous venous return. A variety of views supplemented by color-flow mapping may be necessary to identify the anomalous connection.93
Anomalously connected pulmonary veins may be entered directly with the venous catheter. Selective biplane angiograms in these vessels will document their site of connection. Left-to-right shunting with partial anomalous pulmonary venous connection and an intact atrial septum is usually small or moderate and may go undetected by oximetry techniques. Selective indicator dilution curves in the right and left pulmonary arteries with systemic arterial sampling can detect the lung with the anomalous pulmonary venous connection, and selective biplane angiograms in the pulmonary arterial branches will visualize these connections.
Patients with partial anomalous pulmonary venous connection with ASD appear to follow a course similar, if not identical, to that of patients with an isolated ASD. When the atrial septum is intact, the course depends primarily on the volume of pulmonary venous blood returning to the right side of the heart. Rarely, PVOD may be found even in the presence of a single anomalously connected pulmonary vein and an intact atrial septum.94
Increasing left atrial pressure caused by mitral valve disease or diminished left ventricular compliance will, in the course of time, encourage redistribution of pulmonary arterial blood flow to the portion of the lung drained by the more compliant right atrium. Thus patients who were initially asymptomatic with a modest volume of anomalous pulmonary venous return in youth may become symptomatic and even develop heart failure in adult life.
Asymptomatic patients with small shunts require no treatment. Those with symptoms, larger pulmonary blood flows, congestive failure, or PAH require surgical correction. With an intact atrial septum, precise preoperative identification of the site of the anomalous venous connection is essential. Long-term follow-up in patients who have not had surgery is indicated to detect increasing flow or the appearance of PAH.
Anomalous connection of a right pulmonary vein or veins to the superior vena cava usually is associated with a sinus venosus ASD (see Fig. 83–11; see also Atrial Septal Defect: Surgical Management above). Partial anomalous pulmonary veins draining to the superior vena cava, inferior vena cava, or right atrium are repaired by diversion through the ASD into the left atrium, using a patch baffle. Isolated left-sided anomalous pulmonary veins draining to the left ascending vertical vein or the left superior vena cava are detached and anastomosed directly to the left atrial appendage. Long-term morbidity and mortality are minimal among patients with uncomplicated partial pulmonary venous connections, equivalent to those observed after closure of an ASD.
AV canal defects are characterized by defects in isolation or combination, including an ASD in the lowermost part of the atrial septum (ostium primum), a cleft of the mitral valve (either alone or in combination with a cleft of the tricuspid valve), or VSD. In the most severe form (complete AV canal defect), there is a large ostium primum ASD, a large VSD in the upper muscular septum, and a common AV valve straddling the ventricular septum (Figs. 83–12 and 83–13). The condition appears to result from incomplete growth of the AV endocardial cushions and the AV septum.
Figure 83–12.
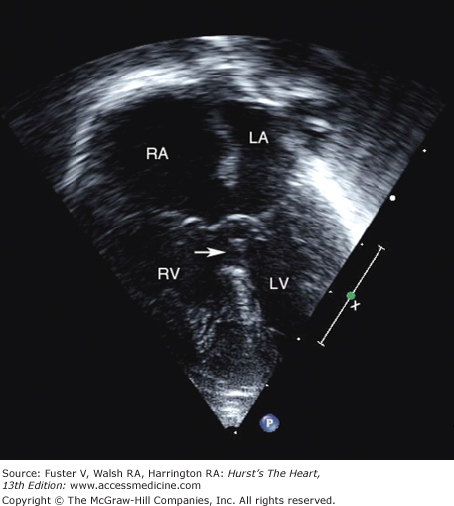
Apical four-chamber echocardiogram view of complete common atrioventricular canal, Rastelli type A. Note the large deficiency of both atrial and ventricular septa as well as the single common atrioventricular valve. The arrow points to the attachment of the superior bridging leaflet to the ventricular septal crest. LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
The ostium primum type of ASD is characterized by a crescent-shaped upper border with no septal tissue forming the lower border. The lower aspect of the defect is bounded by the atrial surfaces of the AV valves and, in the complete type (see Complete Type below), in part by the upper edge of the ventricular septum. A small amount of septal tissue separates the defect from the posterior atrial wall.
Variations occur with respect to the nature of the AV valves. Rogers and Edwards first introduced the terms partial and complete to describe these types.95
The ostium primum ASD is associated with a “cleft” in the anterior mitral leaflet or, probably more accurately, a septal commissure between the superior and inferior leaflets of the left AV valve.77 The tricuspid valve is not cleft or shows a minor central deficiency. The ventricular aspects of the anterior mitral valve elements are fused to the upper edge of the deficient ventricular septum, precluding an interventricular communication. If there is no atrial septal tissue or if the atrial septum is so rudimentary that it produces a common chamber involving both atria, the term common atrium or single atrium is applied.
The complete type of common AV canal is characterized by failure of partitioning of the primitive canal into separate AV orifices. The orifice between the atria and the ventricles is guarded by a common valve with the anterior leaflet derived from the ventral AV endocardial cushion and represents the anterior halves of the anterior mitral and septal tricuspid leaflets. The posterior leaflet originates from the dorsal AV endocardial cushion and represents the posterior halves of the anterior mitral and septal tricuspid leaflets.
Usually, considerable space exists between the anterior and posterior leaflets above and the ventricular septum below; thus in most cases of the complete type, there is free communication between the ventricles.
Rastelli et al96 subdivided the complete variety into three subgroups—types A, B, and C—on the basis of the structure of the common anterior leaflet and its chordal attachments to the ventricular septum and/or papillary muscles (Fig. 83–14; see also Fig. 83–13). Considering the posterior common leaflet, there is variation among the three types in regard to the presence or absence of subdivision and whether the posterior leaflet is attached to the ventricular septum by chordae or by an imperforate membrane.
Variations from the classic types of AV canal defects are recognized, the most common being the AV canal type of isolated VSD, isolated ostium primum ASD without malformed AV valves, and isolated cleft of the anterior mitral or septal tricuspid valve leaflets.
In the asplenia syndrome, the complete variety is almost universal; with polysplenia, it occurs in approximately one-quarter of cases.97 An ASD of the secundum type is present in approximately half of these cases. A double orifice of the mitral valve may be associated with the incomplete type, and tetralogy of Fallot may be associated with the complete type.
If the communication at the ventricular level is large, the right ventricular and pulmonary artery pressures will be elevated. These patients are similar to those with large VSDs. Patients with a communication at the atrial level usually have only normal or slightly elevated systolic pressures in the right side of the heart and a large pulmonary blood flow, as in the secundum type of ASD. Defects in the tricuspid valve, mitral valve, or both may result in severe regurgitation or direct shunting of blood from the left ventricle to the right atrium.
Approximately 3% of infants and children with congenital heart disease have AV canal defects. The majority, some 60% to 70%, have the complete form, with the female-to-male ratio being approximately 1.3:1. Well over half of patients with the complete form have associated Down syndrome. Among children with Down syndrome, 45% have some form of congenital heart disease, with malformations of the type involving the AV canal comprising approximately 50% of these abnormalities.98
Only if the mitral valve is incompetent do the symptoms of patients with partial AV canal differ from those associated with a secundum type of ASD. The complete form of AV canal or the partial form connected with significant mitral regurgitation may be associated with poor weight gain, easy fatigue, tachypnea, repeated respiratory infections, and congestive heart failure. Patients with complete AV canal are almost invariably very sick.
The findings with a partial defect are those of an ASD. If the cleft anterior mitral leaflet is incompetent, the findings of mitral regurgitation also will be present. The physical findings with the complete AV canal defect are those of a very large VSD, usually with full-blown congestive failure. The murmur of mitral regurgitation may not be heard or recognized as such.
Overall cardiac enlargement that is out of proportion to the degree of pulmonary plethora or a cardiac silhouette, suggesting combined ventricular dilatation, may serve to distinguish an uncomplicated secundum ASD from a primum defect with significant mitral regurgitation. Marked cardiac enlargement and severe pulmonary overcirculation are features of the complete AV canal defect.
One of the most helpful diagnostic features in distinguishing individuals with AV canal defects from those with isolated ASDs or VSDs is the characteristic superior orientation of the mean QRS axis in the frontal plane, with a right bundle-branch delay in the precordial leads. Between 92% and 95% of both types of canal have a QRS axis lying between 0 and 150 degrees. The patterns of atrial and ventricular hypertrophy reflect the underlying hemodynamic abnormalities.
Two-dimensional echocardiography is capable of visualizing the extent of septal defects and, with Doppler study and color-flow mapping, left-to-right shunting at the atrial and/or ventricular level, and associated mitral and/or tricuspid valvular regurgitation. The anatomic features of the anterior AV leaflet and its connections may be visualized with sufficient clarity to permit subdivision of complete AV canal defects into types A, B, and C (see Fig. 83–13). Straddling AV valves, a double-orifice mitral valve, single papillary muscles, and hypoplasia or outflow obstruction of the right or left ventricle also can be determined with this technique.99
Figure 83–13.

En-face echocardiogram view of complete common atrioventricular canal, Rastelli type A. En-face imaging from sub-xiphoid view shows the multiple leaflets of the common atrioventricular valve; note the attachment of the superior bridging leaflet to the ventricular septum (arrow). RV, right ventricle.
Cardiac catheterization is rarely performed if the echocardiogram is characteristic and if the history, clinical examination, and echo suggest a large left-to-right shunt and low pulmonary resistance. When it is performed, a significant increase in oxygen saturation between the superior vena cava and the right atrium is present. A right ventricular or pulmonary arterial systolic pressure in excess of 60% of the systemic systolic pressure favors the presence of a complete canal. With a large communication between the two ventricles below the AV valves, the right ventricular, pulmonary arterial, and systemic arterial systolic pressures are virtually identical. Left ventricular angiography in the frontal view demonstrates the “gooseneck deformity” of the left ventricular outflow tract that is characteristic of AV canal malformations and allows a semiquantitative assessment of the degree of mitral regurgitation and shunting from the left ventricle to the right atrium. The left anterior oblique view with craniocaudal angulation is recommended for visualizing the interventricular defect and judging the extent of ventricular septal deficiency. Aortography will confirm the presence of a PDA.
Partial defects without significant mitral regurgitation follow a course similar to that described for the secundum type of septal defects. Moderate or severe mitral regurgitation produces heart failure with resulting symptoms and growth retardation. Infants with a complete AV canal without protective pulmonary stenosis quickly develop and continue to have congestive failure until the course is altered by death, the development of PVOD, or surgical intervention.
Children with an uncomplicated partial defect are managed in the same manner as children with an uncomplicated ASD. Those who are symptomatic should undergo early surgical closure of the primum ASD and plication of the cleft in the septal commissure of the left AV (“mitral”) valve. The few patients with significant residual mitral regurgitation after surgery are managed medically until mitral valve replacement is appropriate. Those without symptoms are repaired before they start school, with the recent trend favoring intervention within the first 2 years.
The approach to an infant with complete AV canal is the same as that for an infant with a large VSD but is tempered by the knowledge that spontaneous improvement is very unlikely except at the expense of the pulmonary vascular bed. Repair is recommended by 6 months of age or earlier with intractable heart failure. Elevation of pulmonary vascular resistance in the first year of life warrants surgical intervention without delay.
With regard to genetic counseling, the risk of a subsequent sibling having heart disease in the presence of a single affected family member is in the range of 2%; it is probably higher for the offspring of an affected parent, particularly if that parent is the mother.100 Concordance for AV canal defects among affected siblings or offspring is much higher than it is with other forms of congenital heart disease and approaches 90%.
The remarkable clinical improvement that follows anatomic repair of complete common AV septal defects in infancy encourages correction early in the first year of life.101 Banding of the pulmonary artery in a critically ill infant with a large ventricular defect was used in the past but has been replaced by operative repair in most centers. The specifics of repair are dictated by anatomic detail: individual variation is considerable (see Fig. 83–14), but the creation of a competent, nonstenotic left-sided AV (“mitral”) valve is essential for an acceptable early and long-term prognosis.
Figure 83–14.

Steps in the repair of the complete form of common atrioventricular canal, type A. A. and B. A pericardial patch is sutured to the ventricular septum. C. and D. The anterior leaflet of the mitral valve is reconstructed and attached to the patch. A portion of the tricuspid leaflet is attached to the patch. Reproduced with permission from Rastelli et al.86
A patch is usually sutured to the right side of the ventricular septum to obliterate the interventricular communication. The anterior and posterior components of the common valve are divided, and the mitral valve is sutured to the patch at an appropriate level. The “cleft” between the left anterior and posterior leaflets is sutured to provide competence without the creation of stenosis. Prosthetic valve implantation rarely is required during primary anatomic repair.102 The right-sided AV (“tricuspid”) apparatus, although less critical to survival, is repaired using the same principles. The interatrial communication is usually closed with a separate piece of pericardium to minimize hemolysis in the presence of residual mitral regurgitation.102 Mitral valve competence is assessed by gentle distension of the left ventricle with cold saline and, more recently, transesophageal echocardiography.
A partial AV canal is repaired through a right atriotomy. The cleft may be closed with a few simple interrupted sutures to encourage inversion and coaptation of the leaflet margins. The ASD usually is closed with a pericardial patch.
Permanent complete heart block once contributed substantially to early mortality and morbidity but is now rare. Patients undergoing repair of a partial AV canal should be observed for the possible development of subaortic left ventricular outflow tract obstruction caused by redundant or residual endocardial cushion tissue.
In-hospital mortality after correction of a complete AV canal in infancy ranges from 3% to 10%103,104; the highest mortality is encountered during the first few months of life in infants with severe AV valve regurgitation, elevated pulmonary vascular resistance, hypoplasia of the left or right ventricle, or other cardiac malformations. At Children’s Hospital in Boston, 191 children with a median age of 4.6 months were repaired between January 1990 and December 1998, with an operative mortality of 1.5%. Reoperation was necessary in 22 patients (11.7%) at a mean of 20 months later—18 for residual mitral regurgitation and 4 for left ventricular outflow tract obstruction.105 Successful correction of a complete AV canal can be accomplished despite associated tetralogy of Fallot, double-outlet ventricle, and other complex anomalies.102
Extracardiac Communications between the Systemic and Pulmonary Circulations, Usually Without Cyanosis
Patent ductus arteriosus, the most common type of extracardiac shunt, represents persistent patency of the vessel that normally connects the pulmonary arterial system and the aorta in a fetus (Fig. 83–15).
Figure 83–15.
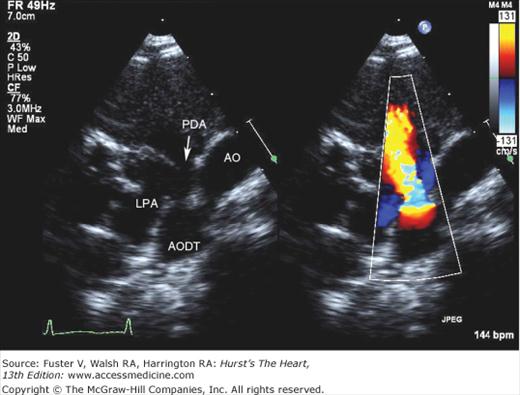
Patent ductus arteriosus (PDA; arrow). Suprasternal echocardiogram “ductal” view in this infant with a large PDA demonstrates left-to-right flow across the vessel (red). Normal antegrade blue color flow is noted in the left pulmonary artery (LPA). AO, aorta; AODT, descending thoracic aorta.
The ductus arteriosus usually closes within 2 or 3 days after birth and becomes the ligamentum arteriosum, but it may remain patent for several months before spontaneous closure. It courses from the origin of the left pulmonary artery below to the lower aspect of the aortic arch just beyond the level of origin of the left subclavian artery above. The recurrent branch of the left vagus nerve circles around its lateral and inferior aspects. Constriction of the ductus postnatally involves a complex interaction of increased partial pressure of oxygen, decreased circulating prostaglandin E2 (PGE2), decreased PGE2 ductal receptors, and decreased pressure within the ductus. Subsequent vessel-wall hypoxia of the ductus promotes further closure through inhibition of prostaglandin and nitric oxide within the ductal wall.106 Exogenous PGE1 has been used extensively to keep the ductus open postnatally,107 and indomethacin, a prostaglandin inhibitor, can close the ductus in many premature infants in whom persistent patency is disadvantageous.108
Patients with PDA may be divided into groups according to whether the vascular resistance through the ductus is low, moderate, or high. The resistance of the ductus is related not only to its cross-sectional area, but also to its length. In patients with a very small ductus that offers high resistance, the flow across the ductus is relatively small. The extra volume of work on the left ventricle is small, and the pulmonary pressure and resistance are not elevated. Patients with only moderate resistance in the ductus have some increase in pulmonary artery pressure, with a moderately greater volume of shunting across the ductus.
In patients with a large patent ductus, the aorta and pulmonary artery are essentially in free communication; the systolic pressure in the pulmonary artery is equal to that in the aorta. Left ventricular volume overload results from recirculation through the lungs, with pulmonary congestion resulting from increased pulmonary flow and/or left ventricular failure. The left ventricle compensates by dilation, followed in many cases by hypertrophy, and the pulmonary vasculature may respond to the high pressure (see Pulmonary Arterial Hypertension above). The right ventricle is subjected to a pressure load.
If the pulmonary resistance equals or exceeds the resistance of the systemic circulation, there is right-to-left shunting from the pulmonary artery to the aorta, resulting in hypoxemia, especially in the lower body and legs.
The history of the mother’s pregnancy and of perinatal events may provide clues associated with a high incidence of PDA, such as exposure to rubella in the first trimester in a nonimmunized mother. PDA is also more common in premature infants, especially those with birth asphyxia or respiratory distress.109
Symptoms are usually restricted to patients with large shunts that produce heart failure or with other complicating problems, such as respiratory distress in a premature infant. The symptoms related to heart failure have been discussed above. Heart failure is most likely to develop in the first few weeks or months of life. Growth may be affected in those with large shunts and failure. The clinical presentation in a premature infant is usually very different from that in a full-term infant, particularly in one with a birth weight less than 1.5 kg (3.3 lb), who is more likely to have moderate to severe respiratory distress. In these infants, the clinical features of respiratory distress occur very early after delivery, requiring mechanical ventilatory support. The inability to wean from the ventilator over several days or initial improvement followed by increasing ventilatory or oxygen requirements are signs that a PDA may be aggravating the clinical status.
In a full-term infant or child with PDA, peripheral pulses are generally brisk and bounding, especially with the larger shunts secondary to runoff from the aorta to the pulmonary artery in diastole. A patient with elevated pulmonary vascular resistance and a right-to-left shunt will have differential cyanosis, with cyanosis and clubbing of the toes but not the fingers, related to shunting of hypoxemic blood from the pulmonary artery into the descending aorta. The apical impulse may be increased or displaced in those with large left-to-right shunts. The right ventricular impulse is increased in infants and children with significant pulmonary hypertension. The second heart sound may be difficult to hear because of the continuous murmur, but it is usually normal. The pulmonary component is accentuated in those with pulmonary hypertension. The typical murmur is a continuous or “machinery” murmur, best heard at the left upper sternal border and below the left clavicle. It is usually a coarse murmur peaking at or near the second heart sound. In patients with at least a moderate shunt, there is a mid-diastolic rumble at the apex resulting from increased flow across the mitral valve.
Findings on chest roentgenography are also dependent on the magnitude of the shunt. In patients with a small shunt, the chest x-ray is normal. With larger shunts, the left atrium and left ventricle are enlarged. Increases in pulmonary arterial flow on radiograph parallel the magnitude of the shunt. In the presence of heart failure, there are signs of pulmonary edema. In older patients who have developed Eisenmenger physiology, the only abnormality may be marked prominence of the central pulmonary arteries, with rapid tapering to the periphery of the lung fields.
With a small shunt the ECG is normal. Left ventricular hypertrophy of the volume overload type, with deep Q waves and increased R-wave voltage in the left precordial leads, is noted with increasing shunt size with left ventricular volume overload. Right ventricular hypertrophy is seen with pulmonary hypertension.
In medium to large shunts, there is left atrial enlargement, as well as increased left ventricular end-diastolic dimension and mean velocity of circumferential fiber shortening. Shunts can be detected readily with color-flow Doppler imaging of the pulmonary artery (see Fig. 83–15). In those with elevated pulmonary vascular resistance, bidirectional or right-to-left flow is present. Occasionally, a trivial amount of flow is seen through the ductus as an incidental finding in those with or without associated heart disease.
In patients with typical, uncomplicated PDA, cardiac catheterization is unnecessary unless closure in the catheterization lab is contemplated. When catheterization is performed, the catheter usually passes preferentially from the pulmonary artery across the PDA into the descending aorta except when the ductus is very small. The saturation is increased in the pulmonary artery compared with the right atrium and ventricle to a degree relative to the size of the shunt. The pulmonary arterial and right ventricular pressures are elevated in those with a large ductus. The pulmonary vascular resistance is elevated in older patients who have developed changes in the pulmonary vascular bed. These patients also have diminished saturation in the descending aorta once the pulmonary resistance reaches a level that will reverse the shunt. Aortography will opacify the ductus and pulmonary arteries.
The complications related to PDA include infective endarteritis, heart failure, and pulmonary hypertension with vascular damage. Infection of the ductus is a risk regardless of ductal size, increasing with the length of survival. The development of a mycotic aneurysm has the potential to compress the recurrent laryngeal nerve, embolize septic material to the lungs, or rupture. Calcification of the ductal wall is common in adults. In patients with large shunts, heart failure can cause significant morbidity and mortality, particularly in a premature and young infant, and sudden death can occur. Progressive damage to the pulmonary vascular bed can occur in some, but it rarely occurs to an irreversible degree in the first year of life. Once irreversible damage occurs, premature death in late adolescence or early adulthood can be anticipated.
With improved echo technology, children without associated heart disease are identified with a trivial amount of flow through a very small (<1 mm) patent ductus. Frequently, the shunt is too small to produce an audible murmur. The natural history of this echo-Doppler–discovered ductus arteriosus without clinical findings is unknown, but most think it is benign as cardiologists have not noted patients with endarteritis in a “silent” ductus.
Interruption of flow through the PDA is the goal of management. For those in heart failure, usually infants, medical management with diuretics and fluid restriction may play a role, but the ultimate aim in infants is closure to prevent heart failure, reduce respiratory insufficiency, and promote growth, whereas with intervention in older children, prevention of endarteritis and pulmonary vascular disease is paramount.
For premature infants, treatment with indomethacin is usually the first-line therapy.110 Successful closure depends on both the dosage and the timing of treatment, although the major determinants seem to be birth weight and gestational age. Because of ductal recurrence, serial treatment regimens may be necessary, especially in those weighing <1000 g at birth. There is increasing evidence that the administration of “prophylactic” indomethacin in infants weighing <1000 g at birth may be associated with a higher closure rate and a better outcome, but reopening of the ductus still occurs frequently.111 Indomethacin therapy has been associated with an increased bleeding tendency resulting from platelet dysfunction, decreased urine output secondary to renal dysfunction, and necrotizing enterocolitis.108 Ibuprofen has achieved closure rates equivalent to those of indomethacin with less renal toxicity,112 but questions remain regarding the potential for chronic lung disease and pulmonary hypertension.
For premature infants when the PDA fails to close with indomethacin or for term infants with a persistent PDA, surgical or interventional closure is recommended. If the PDA is large, there is usually a substantial left-to-right shunt. In these infants, the indication for closure is heart failure, often with failure to thrive. Even in the absence of these indications, when a large PDA is associated with PAH, closure is recommended to prevent PVOD. In children with a smaller PDA and an audible murmur but no evidence of hemodynamic embarrassment, closure has been recommended historically because of the incidence of bacterial endarteritis, in the range of 30% over a lifetime. For children with a PDA but without a heart murmur discovered incidentally when an echocardiogram is performed for other reasons, the need for closure remains controversial.
Surgery for a persistent PDA was first reported more than 60 years ago. The safety and efficacy of this procedure, even in very young children, are well established, with risks that are very low (well under 1%), with success of interruption almost universal. The PDA is exposed and mobilized through a small left thoracotomy in the fourth intercostal space. Ductus obliteration is accomplished by division or ligation. A short, broad, or thin-walled ductus is divided between vascular clamps. The ends are closed with a continuous suture. A long, narrow, thick-walled ductus can be divided or ligated with the sutures at each end anchored superficially in the ductal wall to prevent migration and assure thrombosis and obliteration.
The fragile, thin-walled PDA of a premature infant is obliterated by gentle ligation with a thick suture to minimize disruption or, if small, by occlusion using metallic surgical clips. Some surgeons prefer extrapleural exposure. Ligation in the neonatal intensive care unit, avoiding transport to the operating room, is performed in some centers. Ductus obliteration offers clinical improvement in infants weighing as little as 500 g, with minimal operative risk, a reduced incidence of necrotizing enterocolitis, a reduced duration of intubation, and improvement in late survival.113
Closure of a PDA in an adult requires particular caution; calcification and rigidity of the ductus wall complicate clamping. Placement of a Dacron patch over the aortic orifice of the ductus from within the aorta may be advisable.114
Recently, less-invasive video-assisted thorascopic surgery has been used for PDA closure.115 At Children’s Hospital in Boston, this procedure has been applied to premature infants as small as 575 g, with discharge from the hospital the day after the procedure in full-term infants and children.116
The PDA can be closed by interventional catheterization techniques. In 1971, Portsmann and Wierny introduced a rather complex methodology to plug a PDA by using a transarterial and transvenous approach employing very large catheters.117 Subsequently, Rashkind and Cuaso introduced the use of a double-umbrella device to plug a PDA,118 but the large size of the delivery sheath of the Rashkind device made it inapplicable for small children. Gianturco coils—thin metallic wires glossed with Dacron that assume a coil configuration when released from a catheter—have become an attractive alternative, as they can be delivered through relatively small catheters and have been found to be quite effective, although their usefulness is limited to those older than 8 months of age with PDAs that are no >3.5 or 4.0 mm at the narrowest point.119 Results have been very promising, with a 90% success rate. Newer devices have been introduced that can occlude PDAs of larger luminal caliber, including the Amplatzer ductal occluder (Fig. 83–16).120
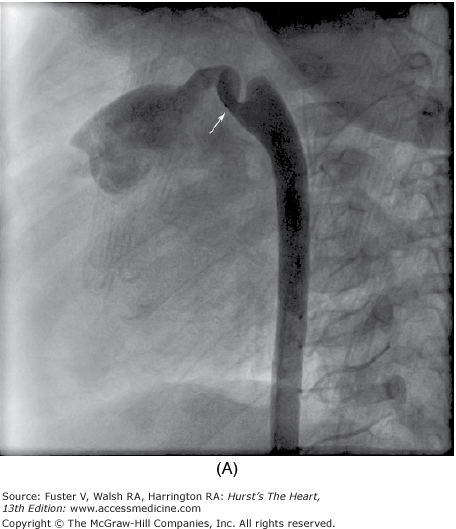
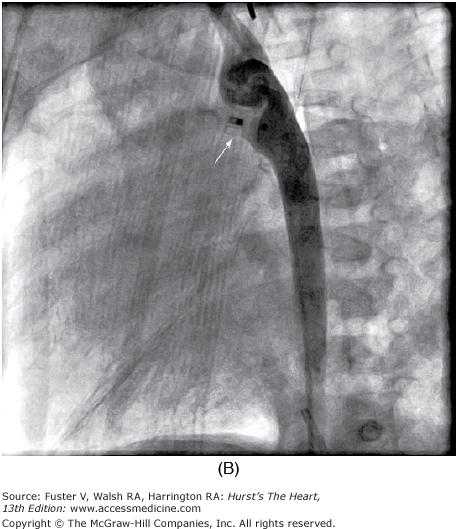
With several highly successful, low-risk, inexpensive, and minimally traumatic procedures available to close a persistent PDA in a neonate, child, adolescent, or adult without pulmonary vascular disease, local experience should guide the preferred option in an individual child.
Sinus of Valsalva fistula is uncommon; it also is referred to as aortic sinus aneurysm. Because of an assumed intrinsic weakness at the union of the aorta with the heart, the aortic media may separate from the aortic annulus and retract upward. The structure that lies between becomes aneurysmal and may rupture to form a fistula. The usual sites of the defects are the posterior (noncoronary) sinus aneurysms that rupture through the atrial septal wall into the right atrium and those of the right sinus that rupture into the right ventricular infundibulum.121 The aneurysm is represented by a colored pouch with multiple perforations in the wall. The principal associated condition is a supracristal VSD in cases with aneurysms of the right sinus (approximately 50%).
Sinus of Valsalva fistulas are most common in adults.122 When the rupture is secondary to bacterial endocarditis, evidence of a preceding infection is found. If the rupture occurs slowly, a small fistulous tract into the right atrium or ventricle develops and presents as the new onset of a small left-to-right shunt. With sudden rupture, there is usually a tearing pain in the mid-precordium associated with dramatically rapid development of pulmonary congestion caused by the sudden onset of a large shunt. Characteristically, the murmur is loud and continuous but is heard lower on the chest than is the murmur of PDA. A to-and-fro murmur rather than a continuous one may be heard at times. The apical impulse is hyperdynamic, and the pulse pressure is widened. VSD may complicate the clinical picture. Echocardiography or cardiac catheterization will confirm the level of the shunt. A pressure difference across the right ventricular outflow tract may be present if the right sinus is involved. Aortography or Doppler echocardiography123 will confirm the diagnosis.
With slow rupture and a small shunt, the major risk is infective endocarditis or extension of the rupture with an increasing shunt. With a large shunt, the heart failure is usually rapidly progressive and may result in a quick demise. A few patients seem to stabilize in this situation.
Appropriate cultures should be drawn and antibiotics started if endocarditis is suspected. Treatment of heart failure should be instituted rapidly. Because of the natural history, all patients should have this condition corrected surgically.
Aneurysms or fistulas from the noncoronary or right coronary sinuses are repaired through the aortic root while the patient is supported on total cardiopulmonary bypass with moderate hypothermia, using techniques similar to those employed for aortic valve replacement. The aortic valve leaflets, the margins of the aneurysm, and the coronary arterial orifices must be visualized precisely. Aneurysms of the noncoronary sinus can be repaired through the right atrium; those arising from the right coronary sinus are accessible through the right ventricle. In most cases, the orifice of the aneurysmal fistula is surgically obliterated using a Dacron patch. In a recent series of 129 patients, reparative methods included plication (47%), patch repair (40%), and aortic root replacement (12%). Sixty percent of those patients needed aortic valve replacement at the same time.122
A conal, or supracristal (type I), VSD must be sought and closed through either the aortic valve or the right ventricular outflow tract when an aneurysm of the right coronary sinus extends into the right ventricle. Surgical results are usually quite good. In the large series cited above, the operative survival was 96%, with no late deaths in an average of 5.9 years of follow-up.122
Though the experience is limited, one center has successfully closed these lesions in the catheterization laboratory using an Amplatzer duct occluder or Gianturco coils. The procedure was successful in closing the defect without residual VSD or aortic regurgitation.124
Valvular and Vascular Malformations of the Left Side of the Heart with Right‐to-Left, Bidirectional, or No Shunt
Coarctation of the aorta is a discrete narrowing of the distal segment of the aortic arch. The characteristic lesion is a deformity of the media of the aorta that involves the anterior, superior, and posterior walls and is represented by a curtain-like infolding of the wall that causes the lumen to be narrowed and eccentric.125 In infants, the lesion lies either opposite the ductus or in a preductal location. In adolescents and adults, it is usually at the ligamentum arteriosum. An aberrant right subclavian artery may be associated. In rare cases, the narrowing lies proximal to the origin of the left common carotid artery or involves a segment of the abdominal aorta. The principal cardiac abnormality is left ventricular hypertrophy. In some infants, left ventricular endocardial fibroelastosis may be associated. Tubular hypoplasia of the distal aortic arch and isthmus is very common, especially with associated cardiac abnormalities involving left heart obstruction.126 The proximal aorta may show a moderate degree of cystic medial necrosis. Beyond the coarctation, the lining may show a localized jet lesion. Prominent collaterals are characteristic in older infants, children, and adolescents. They may be divided into anterior and posterior systems, with the anterior system originating with the internal mammary arteries and making use of the epigastric arteries in the abdominal wall to supply the lower extremities. The posterior system involves parascapular arteries connected with the posterior intercostal arteries and carries blood to the distal aortic compartment principally for supply of the abdominal viscera. The anterior spinal artery, receiving branches from the proximal and distal compartments of the aorta, is also dilated and tortuous.
The most commonly associated defects are tubular hypoplasia of the aortic arch, PDA, VSD, and aortic stenosis (valvular and/or subvalvular). A bicuspid aortic valve is present in 46% of autopsy cases.125
In most instances, both the systolic and diastolic arterial pressures above the coarctation are elevated. Below the coarctation, the systolic pressure is lower than that in the upper extremities, and the diastolic pressure is usually near or only slightly below the normal range. The mechanism of upper-extremity hypertension appears to involve the increased resistance to aortic flow produced by the coarctation itself, the decreased capacity and distensibility of the vessels into which the left ventricle ejects, and humoral factors.127
Coarctation of the aorta occurs in approximately 4% of all infants and children with congenital heart disease and is the predominant lesion in approximately 8% of infants presenting with critical heart disease in the first year of life. It ranks behind only VSD, D-transposition of the great arteries, and tetralogy of Fallot.3 Among all individuals born with coarctation, approximately half present within the first month or two of life with heart failure. Approximately 50% of infants so admitted have uncomplicated coarctation; the remaining half can be expected to have at least one complicating cardiac abnormality. VSD is the most common (64%), followed by left ventricular outflow tract obstruction (31%).128 The timing of ductal tissue constriction in terms of both ductal closure and perhaps aortic constriction appears to play a decisive role in the onset or worsening of symptoms in most of these patients. The male-to-female ratio is approximately 3:1 for isolated coarctation but is only 1.1:1 for complicated coarctation. Approximately 45% of children with Turner syndrome have coarctation.
The clinical picture in a symptomatic infant is one of tachypnea, difficulty in feeding, and poor weight gain. Older children are for the most part asymptomatic, although a few complain of mild fatigue, dyspnea, or symptoms of claudication of the lower extremities when running.
In a symptomatic infant, signs of heart failure are characteristic. A gallop rhythm is common, and a murmur from associated defects or from the coarctation itself (posteriorly in the interscapular area) may be heard. Frequently, these murmurs are either inaudible or nondescript on admission and become characteristic only when heart failure is controlled. The left ventricular impulse is forceful. An early systolic ejection click at the apex suggests the presence of a bicuspid aortic valve. The murmur from the coarctation is medium-pitched and systolic, best heard posteriorly in the interscapular area, usually with some degree of radiation to the left axilla, apex, and anterior precordium. Low-pitched, continuous murmurs of collateral circulation may be heard over the chest wall, particularly posteriorly, but seldom before adolescence.
A systolic blood pressure difference between the upper and lower extremities may be difficult to appreciate or measure in infants with severe heart failure or with a large VSD or PDA. With improved hemodynamic status, pulses in the upper extremities become readily palpable. The femoral pulses remain weak, delayed, or absent. In these very young infants, it is important to assess the brachial and carotid pulses. Weak or absent pulses in all sites are more characteristic of critical aortic stenosis or aortic atresia.
In older children and adults, the radial arterial pulses are typically strong compared with the femoral arteries. A repeatedly measured systolic or mean pressure difference between the upper and lower extremities >10 mm Hg is diagnostic. The pulse pressure in the leg is reduced, and in some patients no pressure can be measured by auscultation or Doppler. Approximately one-third of older children have hypertension. Some patients have only a mild pressure difference between the arms and the legs at rest but a much larger difference during treadmill exercise. A systolic pressure difference between the two arms suggests that the origin of one subclavian artery is at or below the obstruction, for example, aberrant right subclavian from the descending aorta.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


