Congenital Heart Disease in Adults: Introduction
The incidence of moderate and severe forms of congenital heart disease (CHD) is 6 per 1000 live births. If bicuspid aortic valves (BAVs) are included, the incidence increases to 19 per 1000 live births.1 Without early medical or surgical treatment, the majority of patients with complex CHD would not survive to adulthood.2 Surgical and medical advances over the past 60 years have dramatically altered the once bleak prognosis of patients with CHD. In the current era, more than 85% of patients with CHD survive to reach adulthood, and most live productive and functional lives.3,4 Many patients have undergone surgical interventions that were once thought to be curative. With the exception of early surgical ligation of a patent ductus arteriosus (PDA), a surgical “cure” for CHD without operative sequelae or need for reoperation does not exist.5
Adults with operated and unoperated CHD require long-term follow-up. This long-term care should include cardiologists with experience in or who specialize in adult CHD. Standardized diagnostic and treatment guidelines have emerged from the American College of Cardiology (ACC) and American Heart Association (AHA).6 Diagnostic and treatment errors by cardiologists inexperienced in patients with complex CHD can be minimized by improving understanding of the pathophysiology, hemodynamics, and prognosis of various lesions. Patient referral to regional adult CHD experts is encouraged to facilitate specialized counseling and management. These patients also face a variety of psychosocial issues, including self-image problems; difficulties in acquiring and maintaining health and life insurance; exercise and activity limitations; and issues of sexuality, contraception, and reproduction.
Specialized adult CHD centers have developed in North America and in Europe, but there remains a need for more specialized centers even in these regions.7 Requirements include close collaboration among adult and pediatric cardiologists, cardiac surgeons, nurse specialists, and consultants. Training and research are of pivotal importance. The development of management, research, and training guidelines over the past decade is a major step forward for the field of adult CHD.8-12 Another encouraging development is the establishment of the Adult Congenital Heart Association (ACHA) in the United States. This patient-initiated association is a nonprofit organization that seeks to improve the quality of life and extend the lives of adults with CHD. Multicenter and prospective clinical research in adult CHD have increased in frequency and variety.13 A dedicated multicenter research network, the Association for Adult Research in Congenital Cardiology, was established in 2007 in North America. The goal of this network is to foster and implement multicenter research.14
Medical Considerations
Many adults with CHD have not had, or may never require, surgical intervention. The most common defects incidentally encountered in adulthood are small ventricular or atrial septal defect (ASD), mild pulmonary stenosis, BAVs, and mitral valve prolapse (Table 84–1). Less common, but more complex, conditions include nonrestrictive central shunts with right-to-left shunt reversal resulting in the cyanotic Eisenmenger syndrome. Most patients require long-term follow-up to monitor for disease progression, appropriately treat possible sequelae or complications, and reinforce the need for a healthy lifestyle and diligent dental care.6 Women of childbearing age and men wishing to father children should be informed of the risks of pregnancy and the increased risk of CHD in their progeny. An increasing number of patients who underwent palliative or corrective operations in childhood are reaching adulthood (Table 84–2). Many patients are under the false impression that they are “cured” and are lost to follow-up until symptoms bring them back to medical attention. These patients should be monitored on a regular basis for progression of disease and need for reoperation.6 Uncorrected, corrected, and palliated patients often require repeat operations, interventional catheterizations, and electrophysiologic therapies in adulthood. At least intermittent consultation with an adult CHD center and performance of advanced diagnostic studies and interventions at experienced centers are highly recommended given the potential complexity and rarity of some conditions.6
| 2006 VHD Guideline Recommendations | 2008 VHD Focused Update Recommendations |
|---|---|
| Class I | Class IIa |
Prophylaxis against infective endocarditis is recommended for the following patients: • Patients with prosthetic heart valves and patients with a history of infective endocarditis. (Level of Evidence: C) • Patients who have complex cyanotic congenital heart disease (eg, single-ventricle states, transposition of the great arteries, tetralogy of Fallot). (Level of Evidence: C) • Patients with surgically constructed systemic pulmonary shunts or conduits. (Level of Evidence: C) • Patients with congenital cardiac valve malformations, particularly those with bicuspid aortic valves, and patients with acquired valvular dysfunction (eg, rheumatic heart disease). (Level of Evidence: C) • Patients who have undergone valve repair. (Level of Evidence: C) • Patients who have hypertrophic cardiomyopathy when there is latent or resting obstruction. (Level of Evidence: C) • Patients with MVP and auscultatory evidence of valvular regurgitation and/or thickened leaflets on echocardiography. (Level of Evidence: C) | Prophylaxis against infective endocarditis is reasonable for the following patients at highest risk for adverse outcomes from infective endocarditis who undergo dental procedures that involve manipulation of either gingival tissue or the periapical region of teeth or perforation of the oral mucosa: • Patients with prosthetic cardiac valves or prosthetic material used for cardiac valve repair. (Level of Evidence: B) • Patients with previous infective endocarditis. (Level of Evidence: B) • Patients with CHD. (Level of Evidence: B) • Unrepaired cyanotic CHD, including palliative shunts and conduits. (Level of Evidence: B) • Completely repaired congenital heart defect repaired with prosthetic material or device, whether placed by surgery or by catheter intervention, during the first 6 mo after the procedure. (Level of Evidence: B) • Repaired CHD with residual defects at the site or adjacent to the site of a prosthetic patch or prosthetic device (both of which inhibit endothelialization). (Level of Evidence: B) • Cardiac transplant recipients with valve regurgitation due to a structurally abnormal valve. (Level of Evidence: C) |
| Class III | Class III |
Prophylaxis against infective endocarditis is not recommended for the following patients: • Patients with isolated secundum atrial septal defect. (Level of Evidence: C) • Patients 6 or more months after successful surgical or percutaneous repair of atrial septal defect, ventricular septal defect, or patent ductus arteriosus. (Level of Evidence: C) • Patients with MVP without MR or thickened leaflets on echocardiography. (Level of Evidence: C) • Patients with physiological, functional, or innocent heart murmurs, including patients with aortic valve sclerosis as defined by focal areas of increased echogenicity and thickening of the leaflets without restriction of motion and a peak velocity less than 2 m/s. (Level of Evidence: C) • Patients with echocardiographic evidence of physiologic MR in the absence of a murmur and with structurally normal valves. (Level of Evidence: C) • Patients with echocardiographic evidence of physiological TR and/or pulmonary regurgitation in the absence of a murmur and with structurally normal valves. (Level of Evidence: C) | Prophylaxis against infective endocarditis is not recommended for nondental procedures (such as transesophageal echocardiogram, esophagogastroduodenoscopy, or colonoscopy) in the absence of active infection. (Level of Evidence: B) |
Progressive myocardial dysfunction with resultant heart failure is a leading cause of morbidity and mortality in patients with CHD. Patients may present with left, right, biventricular, or univentricular failure in the face of pressure or volume overload often in the presence of hypoxia secondary to coexistent pulmonary vascular disease. Intrinsic myocardial abnormalities may result in restrictive diastolic properties, leading to a chronic low cardiac output state, volume overload, congestive hepatopathy, ascites, and protein-losing enteropathy. Complications of restrictive cardiomyopathy are often progressive and difficult to manage; cardiac transplantation is the ultimate therapeutic option. Physical examination includes a thorough assessment of volume status with specific attention to the jugular venous filling pressure and waveform. Pulmonary hypertension or ventricular filling abnormalities are often accompanied by an amplified A wave in the jugular venous pulse. Right-sided atrioventricular (AV) valve regurgitation may result in an amplified jugular venous V wave proportional to the severity of regurgitation. Percussion and palpation of the liver to evaluate for pulsatility and degree of hepatomegaly is recommended. Doppler echocardiography is indispensable for noninvasive evaluation of hemodynamics and shunt fractions.15,16 Invasive cardiac catheterization is reserved for patients with inadequate acoustic windows that limit the utility of transthoracic echocardiography or in those in whom pulmonary vascular resistance, shunt fractions, or chamber pressures cannot be gleaned from noninvasive methods and must be measured directly.
Operations to palliate or repair congenital cardiovascular lesions were originally devised to address physiologic issues, specifically to increase or diminish the supply of blood to the pulmonary circulation. The early era of congenital cardiac surgery was marked by giant leaps forward in the physiologic treatment of lesions. For example, patients with pulmonary atresia or a single ventricle underwent placement of an arteriopulmonary shunt, a wave of surgical innovation initiated by the famed Blalock-Taussig shunt, initially performed in 194517 (Fig. 84–1). Another example is the atrial switch operation for correction of complete transposition of the great arteries (TGA; Fig. 84–2) in which deoxygenated blood is redirected by the atrial baffle from the cavae to the left ventricle (LV) and thereafter to the pulmonary arterial circulation. The oxygenated pulmonary venous blood returns to the systemic right ventricle (RV). These operations are successful in diverting blood flow to improve or correct physiology but do not normalize anatomy or optimize hemodynamics. Patients with systemic RVs or those with single ventricles are especially susceptible to deteriorating ventricular function. The heterogeneous morphology and loading conditions for these ventricles suggest that standard indices of ventricular function, namely echo or other imaging-derived ejection fraction, may not be as useful in identifying the highest risk subsets. Elevations of serum brain natriuretic peptide (BNP) may help identify patients with increased ventricular wall tension often accompanied by ventricular enlargement and dysfunction.18 Atrial and ventricular arrhythmias increase in frequency and severity as ventricular function deteriorates and themselves lead to further decreases in cardiac output. Medical and surgical interventions are aimed at preservation of function and prevention of arrhythmias.
Figure 84–1.
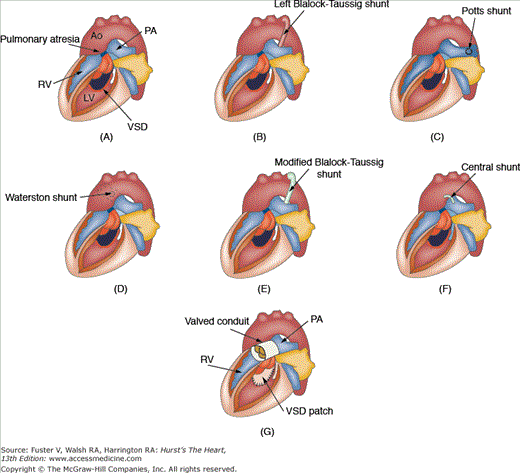
A. Unrepaired tetralogy of Fallot with pulmonary atresia. There is no anterograde flow from the right ventricle (RV) to the pulmonary artery (PA). A large nonrestrictive ventricular septal defect (VSD) allows communication between the left ventricle (LV) and the RV. There is an overriding aorta (Ao). B. Classic left Blalock-Taussig shunt consisting of direct connection of the divided left subclavian artery to the left pulmonary artery. C. Potts shunt consisting of a direct connection between the anterior wall of the descending Ao and left PA. D. Waterston shunt consisting of a direct connection of the posterior wall of the ascending Ao and the right PA. E. Modified Blalock-Taussig shunt consisting of a synthetic (Gore-Tex) tube connection the left subclavian artery to the PA. F. Central shunt consisting of a synthetic tubular connection from the ascending Ao to the PA. G. Intracardiac repair consisting of VSD patch closure and placement of a valved conduit from the RV to the PA.
Figure 84–2.
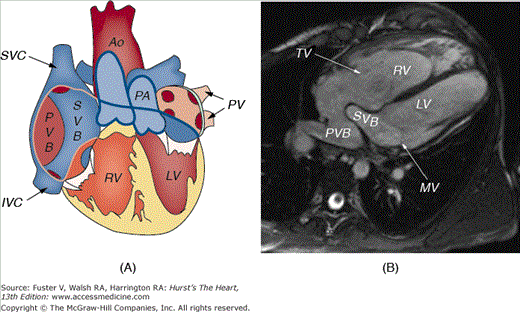
A. Valentine diagram of the cardiac anatomy in a patient with D-transposition of the great arteries who has undergone the Mustard or Senning atrial switch operation. Deoxygenated blood (blue) returning from the superior and inferior vena cavae (SVC and IVC, respectively) is redirected via a systemic venous baffle (SVB) to the left ventricle (LV) and thereafter into the transposed pulmonary artery (PA). Oxygenated blood (red) returning from the lungs via the pulmonary veins (PVs) is redirected via a pulmonary venous baffle (PVB) to the systemic right ventricle (RV) and then to an anterior and rightward aorta (Ao). B. Cine-cardiac magnetic resonance imaging scan (axial plane) demonstrating the SVB directing deoxygenated blood via the mitral valve (MV) to the subpulmonic LV and PVB directing oxygenated blood via the tricuspid valve (TV) to the systemic RV.
Postoperative residual defects may be a major cause of progressive deterioration decades after surgery. Severe chronic pulmonary regurgitation may be well tolerated for decades after surgery.19 Eventually, progressive RV dilatation and elevated filling pressures, particularly if ventriculotomy scars coexist, may create the substrate for ventricular arrhythmias. Medical therapy for heart failure in patients with CHD is adopted from the extensive evidence-based literature in patients with ischemic and nonischemic cardiomyopathies. There is a paucity of prospective randomized data evaluating heart failure therapies in patients with CHD. The renin–angiotensin system may not play as deleterious a role in certain subsets of patients with CHD compared with those with ischemic and nonischemic cardiomyopathies. For example, patients with systemic RVs with depressed systolic function do not demonstrate increased angiotensin levels and therefore do not appear to derive demonstrable intermediate-term benefit from the use of angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs).20 β-Blockers may retard the progression of ventricular dysfunction and decrease the likelihood of arrhythmias.21 Diuretics have not been rigorously evaluated in patients with CHD.
Adults with CHD may be cyanotic because of decreased pulmonary blood flow or admixture of desaturated systemic venous blood with pulmonary venous blood. Decreased pulmonary blood flow may occur in the presence or absence of pulmonary vascular disease. The latter is exemplified by unrepaired tetralogy of Fallot (ToF) with pulmonary atresia in which pulmonary blood flow is predominantly via aortopulmonary collaterals. Cyanosis in the presence of pulmonary vascular disease may be secondary to unrepaired central shunts (Eisenmenger syndrome) or the coincidence of idiopathic pulmonary hypertension in the presence of an ASD.22
In 1897, Victor Eisenmenger described the autopsy findings of a 32-year-old man who had been cyanotic from infancy; findings included a large ventricular septal defect (VSD), pulmonary arteriosclerosis, and pulmonary artery thrombosis. The cause of death was pulmonary hemorrhage. The Eisenmenger complex (the term coined by Maude Abbott) refers to a reversed shunt (right to left) in the presence of a nonrestrictive VSD (Fig. 84–3). Eisenmenger syndrome is a heterogeneous series of lesions leading to a reversed central shunt. During the first few years of life, the small muscular pulmonary artery branches are capable of relaxing, and the defect can be closed with a subsequent gradual decrease in pulmonary vascular resistance. After 2 to 3 years, reactive intimal fibrosis begins to obliterate the lumen of the muscular arteries, and they no longer respond to vasodilating agents such as acetylcholine, adenosine, or nitric oxide.23 This obliterative process represents a fixed obstruction to flow; the shunt reverses, and surgical correction is contraindicated. Patients with trisomy 21 (Down syndrome) are especially susceptible to developing pulmonary vascular disease.24 Patients with cyanosis secondary to pulmonary vascular disease and shunt reversal experience significantly decreased maximal and submaximal exercise capacity compared with other patients with complex CHD (Fig. 84–4).25,26
Figure 84–3.
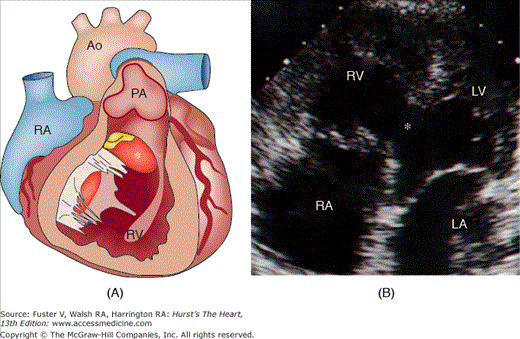
A. Rendering of a heart with Eisenmenger complex, characterized by a nonrestrictive ventricular septal defect (asterisk). The right ventricle (RV) is hypertrophied, and the main pulmonary artery (PA) is dilated and has evidence of atheroma formation. Also labeled are the right atrium (RA) and the aorta (Ao). B. Transthoracic echocardiographic image, apical four-chamber view of a heart with Eisenmenger complex. There is a large nonrestrictive VSD (asterisk). The RV is enlarged and hypertrophied. The left ventricle (LV) and (LA) are labeled.
Figure 84–4.
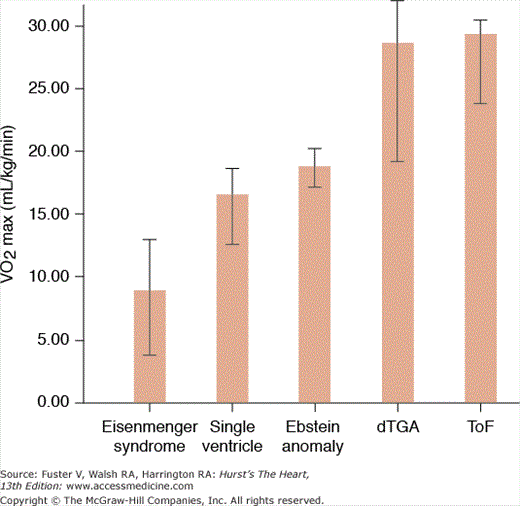
Distribution of maximum oxygen consumption (VO2max) in 78 adults with various types of congenital heart disease followed at the Ahmanson/University of California, Los Angeles, Adult Congenital Heart Disease Center. Surgically repaired patient with tetralogy of Fallot (ToF) and patients with dextro-transposition of the great arteries (dTGA) who have undergone an atrial switch operation performed significantly better than other subgroups. Patients with Eisenmenger syndrome had a mean VO2max of less than 10 mL/kg/min, which was significantly lower than all other subgroups.
Survival in patients with Eisenmenger syndrome is reduced by approximately 20 years compared with healthy control subjects; nearly 50% of patients survive to the sixth decade of life.27 Decreased functional capacity, heart failure, and arrhythmias are predictors of decreased survival. Patients with unrepaired truncus arteriosus and those with single ventricle morphology have a poorer prognosis than patients with nonrestrictive VSD (Eisenmenger complex).28 Patients with truncus arteriosus are at risk of developing truncal valve stenosis and regurgitation; infective endocarditis leading to aortic valve regurgitation is a known complication. With single-ventricle morphology, especially RV morphology, progressive ventricular dysfunction is a concern. Causes of death include pulmonary hemorrhage, pulmonary arterial thrombosis, pulmonary artery dissection, ventricular arrhythmias, and ventricular failure.28 All cyanotic patients are at a heightened risk for infective endocarditis. Interestingly, patients with cyanotic CHD demonstrate an antiatherogenic substrate and are less susceptible to atherosclerotic heart disease than noncyanotic control subjects.29-31 The coronary arterial circulation in cyanotic patients is characterized by markedly dilated and tortuous extramural coronary arteries and a well-developed microcirculation within the myocardium.30,31 Serum lipid levels are low in cyanotic patients and remain low years after correction of cyanosis, suggesting the modulation of as yet unidentified operator genes by cyanosis. The antiatherogenic state of these patients is further characterized by elevated bilirubin levels; increased nitrous oxide production by vascular endothelial cells because of increased shear stress from hyperviscosity; and low platelet levels, incurring less thrombotic risk29,32 Patients with chronic cyanosis also develop defective hemostasis from abnormalities in platelet function and in the coagulation and fibrinolytic systems.33,34 Interestingly, the pulmonary arterial circulation is not spared from atherosclerosis and thrombosis. As a matter of fact, these patients often demonstrate an aggressive atherosclerotic process within the pulmonary arterial tree that correlates with the duration of pulmonary hypertension.35 Cardiac computed tomography (CT) may be used to quantify calcium deposition within the pulmonary arterial tree, calcium being a surrogate marker for atherosclerosis (Fig. 84–5). Histologic examination of necropsy or autopsy pulmonary artery specimens demonstrates a mix of typical calcified lipid-rich atherosclerotic plaques and dystrophic intimal calcification. Pulmonary arterial thrombosis is common and is challenging to treat.36 Thrombi are more common in older patients, those with biventricular dysfunction, and those with decreased pulmonary blood flow velocity.37 Chronic anticoagulation increases the risk of life-threatening pulmonary hemorrhage in patients with thrombocytopenia and should be avoided unless the patient has other definitive indications (eg, atrial fibrillation or deep venous thrombosis).28
Figure 84–5.
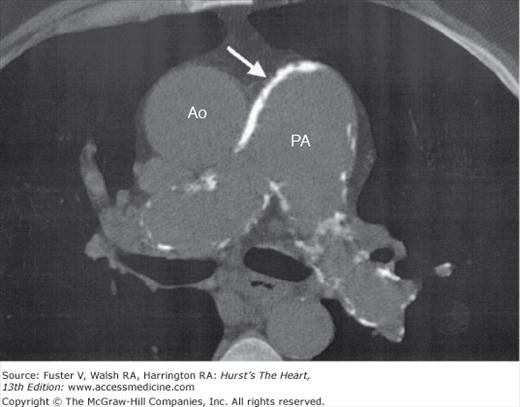
Noncontrast computed tomography scan of a 60-year-old man with Eisenmenger complex demonstrating extensive white calcium deposits (arrow) within the walls of the pulmonary artery (PA) and branch pulmonary arteries. The ascending aorta (Ao) is labeled. Note the relative paucity of calcium in the walls of the aorta compared with the PA.
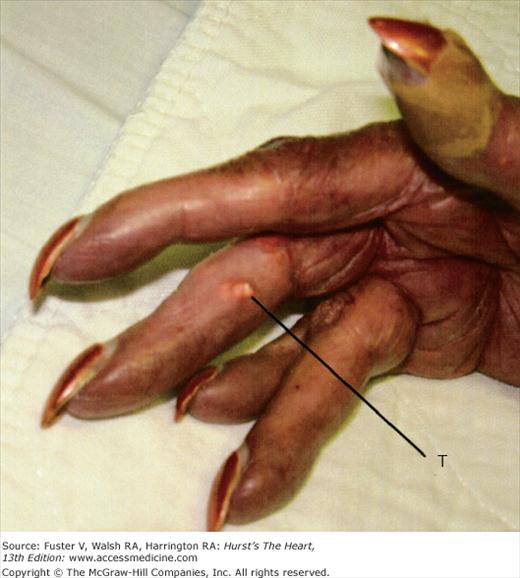
Chronic cyanosis leads to erythrocytosis and may result in hyperviscosity. The risk of symptomatic hyperviscosity is low in patients with a hemoglobin level below 20 g/dL who are not dehydrated. Symptoms of hyperviscosity include headache, dizziness, fatigue, and blurry vision. Judicious phlebotomy may improve these symptoms but should be reserved for patients who do not respond to aggressive hydration.38 Regular phlebotomies should be strictly avoided regardless of the hemoglobin level because of the resultant iron deficiency leading to microcytic and less deformable red blood cells that do not pass through the microcirculation as readily as more deformable iron-replete cells.33 Because of this the paradoxical anemia of erythrocytotic patients with iron deficiency may increase the risk of stroke.34,39 Iron repletion in these patients should be instituted with care because of the tendency for excessive erythrocytosis. Hyperuricemia is common because of increased red blood cell turnover and decreased renal excretion of uric acid; however, urate crystal nephropathy is rare despite the elevated serum uric acid levels. Patients with elevated uric acid levels rarely develop tophaceous deposits within the soft tissue of the elbows or digits; these can be painful and tender (Fig. 84–6). Arthralgia is well recognized, and frank gouty arthritis is rare.40
Patients with cyanosis secondary to right-to-left shunts are at risk for paradoxical emboli from the venous to the systemic arterial circulation, leading to cerebrovascular accidents, renal impairment, or myocardial infarction. Septic emboli may cause cerebral abscesses and must always be considered in cyanotic patients with fever and neurologic symptoms. Air filters should be used with all intravenous (IV) lines, and chronic indwelling venous catheters should be avoided. Anticoagulation may be considered in patients who must have chronic indwelling lines (eg, patients with infective endocarditis who require prolonged IV antibiotic therapy).
Promising advances have occurred over the past decade in the treatment of patients with pulmonary hypertension. The Breathe-5 trial is the first prospective and randomized clinical trial performed solely in patients with Eisenmenger syndrome randomized to either a nonselective endothelin inhibitor (bosentan) or placebo.41 Patients receiving bosentan had a significant decrease in pulmonary vascular resistance and systemic vascular resistance, resulting in increased pulmonary and systemic blood flow. They also had a significant increase in 6-minute walk distance (51 m). Patients were followed for 16 weeks. Long-term follow-up of patients placed on endothelin blockers suggests an attenuation of the benefits over time.42 Phosphodiesterase-5 inhibitors are also effective in the treatment of patients with Eisenmenger syndrome and result in a similar improvement in functional capacity and lowering of pulmonary vascular resistance as endothelin blockade.43 IV prostacyclin use is generally avoided because of the requirement for chronic indwelling venous catheters, which increases the risk of thrombosis and paradoxical embolism; moreover, the presence of a central right-to-left shunt results in much of the IV-administered drug bypassing the pulmonary bed and resulting in systemic hypotension. Inhaled prostacyclin may be more efficacious in this patient population; however, at this time, there is a paucity of data on the use of this agent in patients with Eisenmenger syndrome. Serum BNP is a diagnostically and prognostically important marker in various forms of systolic and diastolic heart failure as well as primary and secondary forms of pulmonary hypertension. Serum BNP is elevated in most patients with Eisenmenger syndrome; however, an outpatient level 250 pg/mL or above in clinically euvolemic patients predicts impending heart failure admission or death.44
Patients with CHD (corrected or uncorrected) are at risk for developing infective endocarditis (see Tables 84–1 and 84–2).45 Certain subgroups are considered at higher risk for infective endocarditis. Guidelines from the European Society of Cardiology and the AHA and ACC on prevention of infective endocarditis place patients with prosthetic valves, cyanosis, and systemic or pulmonary artery conduits, as well as patients with previous endocarditis into a high-risk subgroup.45-47 Most other congenital cardiac conditions are at a moderate risk category, except for patients that have undergone surgical repair of ASD, VSD, or PDA (without residua beyond 6 months) who are considered low risk provided there are no sequelae (eg, aortic valve prolapse, aortic regurgitation). In patients at high risk, antibiotic prophylaxis is recommended for dental procedures associated with significant bleeding from hard or soft tissues, periodontal surgery, scaling, and professional teeth cleaning. Other procedures requiring prophylaxis include respiratory, genitourinary, and gastrointestinal procedures. Poor dental hygiene may produce bacteremia even in the absence of dental procedures. Individuals who are at risk for developing bacterial endocarditis should establish and maintain the best possible oral health to reduce potential sources of bacterial seeding. Optimal oral health is maintained through regular professional care and diligent brushing and flossing on the part of the patient. High-risk patients are advised to use a minimally abrasive soft bristle tooth brush to minimize bleeding. A structured questionnaire mailed to 515 dentists and 85 cardiologists in Ireland revealed that most cardiologists are familiar with the cardiac conditions that pose a risk for dental patients but did not educate their patients on the importance of dental health. On the other hand, dentists were good at identifying dental procedures that place patients at risk but less informed which cardiac conditions warranted prophylaxis, choice of prophylaxis, and appropriate treatment intervals.48 A survey questionnaire of 102 adults with CHD found that patients’ knowledge of their underlying condition, endocarditis risk, and prevention measures was sorely lacking.49
| Hemoglobina (g/dL) | Pregnancy (n) | Live Births (n) | Live Born (%) |
|---|---|---|---|
| ≤16 | 28 | 20 | 71 |
| 17-19 | 40 | 18 | 45 |
| ⩾20 | 26 | 2 | 8 |
| Arterial Oxygenb Saturation (%) | Pregnancy (n) | Live Births (n) | Live Born (%) |
| ≤85 | 17 | 2 | 12 |
| 85-89 | 22 | 10 | 45 |
| ⩾90 | 13 | 12 | 92 |
There is an underrecognized risk of bacteremic infective endocarditis in patients who acquire tattoos or body piercings.50 Patient education is of paramount importance, and patients should carry an information card with them that clearly identifies their endocarditis risk category.
The symptoms of infective endocarditis are often subtle and nonspecific, including low-grade fever, malaise, fatigue, and headache. The diagnosis of endocarditis should be entertained in any patient with unexplained fever or malaise and should prompt blood cultures and echocardiographic imaging. Echocardiography is an integral tool in the diagnosis and follow-up of patients with infective endocarditis.51 The use of transesophageal echocardiography (TEE) is recommended in cases with high clinical suspicion and equivocal or negative findings on transthoracic echocardiography or when a prosthetic valve or conduit is involved52 (Fig. 84–7). Injudicious use of antibiotics without prior blood cultures often makes the culprit organism more difficult to identify by culture, making appropriate treatment more difficult.
Figure 84–7.
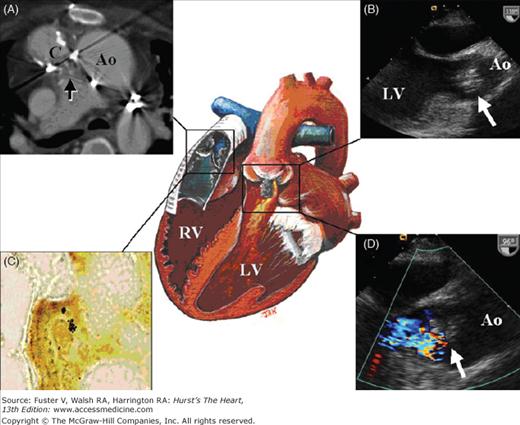
Illustration of a 36-year-old woman with dextrocardia, a ventricular septal defect (VSD), double-outlet right ventricle, and pulmonary atresia. At 13 years of age, she underwent corrective surgery using the Rastelli procedure with baffling of the left ventricular (LV) blood via the VSD to the aorta and placement of a 22-mm valved Dacron conduit from the right ventricle to the main pulmonary artery. The right ventricle (RV) is connected to the pulmonary artery via a valved Dacron conduit. Note the grayish vegetation within this structure. The LV is the systemic ventricle. A baffle directs LV outflow to the aortic valve. The aortic valve has grayish vegetation on it that prolapses into the LV outflow tract and causes aortic regurgitation. A. CT angiogram at the level of the distal RV to pulmonary artery conduit (C). The arrow points to the grayish vegetation within the conduit. The ascending aorta is labeled (Ao). B. Transesophageal echocardiogram demonstrating the vegetation (arrow) prolapsing in diastole from the Ao to the LV. C. Warthin-Starry stain of the grayish vegetation demonstrates a cluster of Bartonella henselae that appear as black specks. D. Transesophageal echocardiogram with color-flow Doppler demonstrating aortic regurgitation around the area of vegetation. The arrow points to the vegetation.
Arrhythmias and conduction defects are common in operated and unoperated patients with CHD and have a major impact on survival and quality of life. Although the principles for diagnosis and treatment of arrhythmias are similar to those used in patients with anatomically normal hearts, there are some notable exceptions. Atrial rhythm disturbances that may be benign and well tolerated with a rate control strategy in a structurally normal heart may be poorly tolerated in complex CHD. Patients with functional single-ventricle or tricuspid atresia who have undergone total cavopulmonary (Fontan) connection exemplify this in that atrial tachyarrhythmias frequently result in deleterious hemodynamics, a decrease in cardiac output, and functional deterioration despite adequate rate control.53 Atrial arrhythmias occur in 15% of adults with CHD. The lifetime incidence increases exponentially with advancing age, and atrial arrhythmias affect more than 50% of adults with complex CHD.7,54,55 Bradyarrhythmias are common in certain subsets of patients, such as those with congenitally or surgically corrected TGA. Special consideration must be given to the use of therapies that have negative chronotropic and inotropic properties. The clinical significance of arrhythmias depends greatly on the hemodynamic context in which they occur.
Abnormalities of sinus node function are common in adults with CHD, especially in patients who have undergone atrial surgery. Patients with surgically corrected ASD, specifically those with sinus venosus ASD, are at a heightened risk of developing postoperative sinus node dysfunction (SND) because of the proximity of the ASD to the sinus node.56 Patients with uncorrected atrial shunts causing chronic right atrial volume overload and right atrial stretch undergo a process of electrical remodeling with increases in effective refractory period, conduction delay at the crista terminalis, and SND. Conduction delay at the crista terminalis persists beyond ASD closure and may contribute to the long-term atrial arrhythmia substrate in this condition.57 Patients who have undergone surgeries that involve extensive atrial reconstruction such as the Senning or Mustard atrial switch operation for complete TGA (see Fig. 84–2) are particularly prone to SND. A retrospective follow-up study of 137 patients who had undergone the Mustard or Senning operation demonstrated that nearly 50% of patients had SND; however, the presence of SND did not influence mortality.58 Frequency of SND is also increased after surgery for correction of ToF, placement of cavopulmonary shunt (Glenn or Fontan), and numerous other congenital heart operations. High-grade AV node block is a well-recognized problem in patients with unoperated congenitally corrected TGA; many patients are initially diagnosed in adulthood when they present with signs and symptoms of bradycardia from high-grade heart block. Injury to the AV node and ventricular conduction tissue may result from surgery for lesions such as VSD, ToF, and mitral or tricuspid valve repair or replacement. Transient complete AV block in the postoperative period has been shown to have prognostic significance, especially if the induced block is below the bundle of His.59 Postoperative right bundle branch block is frequent after right ventriculotomy and is likely secondary to transection of distal Purkinje fibers and postsurgical scarring creating an area of slowed conduction and a substrate for reentrant ventricular arrhythmias.60,61 Although bradycardia has been postulated as the cause of death in some conditions, evidence indicates that tachyarrhythmia is the more likely culprit. However, the presence of underlying bradycardia as a substrate for initiation of tachycardia is well recognized.62 Indications for pacemaker implantation in asymptomatic patients with SND or conduction system disease are controversial because the arrhythmia is benign in the majority of cases. Patients who are asymptomatic at rest or with low levels of activity may become more fatigued and dyspneic with greater levels of exertion. Electrocardiographic (ECG) stress testing identifies patients with chronotropic incompetence leading to decreased exercise capacity.
Pacemaker implantation may be challenging in patients with complex underlying or postoperative anatomy.63 The choice of pacemaker depends on the specific indication. In patients who have isolated SND, a single atrial lead should suffice. Patients who require chronic ventricular pacing often develop progressive cardiomyopathy and ventricular dyssynchrony if a single ventricular pacing is performed; a problem that may be ameliorated by multisite ventricular pacing.64
Tachyarrhythmias occur in patients with operated and unoperated forms of CHD. Atrial tachyarrhythmias are common in patients who have undergone atrial surgery (eg, Senning or Mustard operations) and those in whom AV valve disease or shunts lead to atrial volume or pressure overload. Scar-mediated reentrant atrial flutter is a common theme in the majority of patients who have undergone some form of atrial reconstruction. The various modifications of the Fontan operation have been necessitated by the need to decrease the very high incidence of poorly tolerated atrial tachyarrhythmias seen with the right atrial to pulmonary artery Fontan repair that was initially performed in 197153,65 (Fig. 84–8). Medical or electrical cardioversion should be carried out promptly to restore sinus rhythm; antiarrhythmic medications may be necessary to help maintain it. Surgical cryoablation and transvenous catheter ablation aided by three-dimensional electroanatomic mapping can be used as a therapeutic intervention in atrial tachyarrhythmias of various causes.66-68 Electroanatomic mapping and radiofrequency ablations in patients with CHD represent some of the most challenging electrophysiologic procedures because of the complex pre- and postsurgical anatomy, chamber dilatation, variable anatomy of the conduction system, and presence of intracardiac scars leading to multiple reentrant circuits. An experienced electrophysiology team, including operators and technicians familiar with the complexity of the arrhythmias and substrates, is key to the successful diagnosis and treatment of patients with CHD.
Figure 84–8.
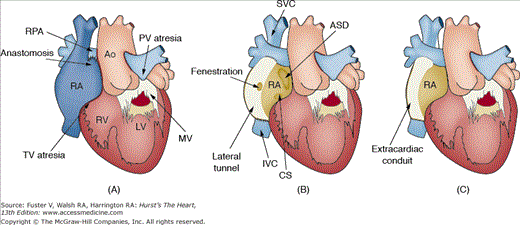
Most commonly used modifications of the Fontan operation. A. Right atrial (RA) to pulmonary artery (PA) Fontan connection with the right atrial appendage directly sutured to the RPA in a patient with tricuspid and pulmonary atresia. The right ventricle (RV), left ventricle (LV), mitral valve (MV), right pulmonary artery (RPA), and aorta (Ao) are labeled. B. The fenestrated lateral tunnel Fontan in which a synthetic material (eg, Gore-Tex) is used to extend a tunnel along the inside lateral wall of right atrium to the RPA, and the superior vena cava (SVC) is sutured directly to the right pulmonary artery. The fenestration in the wall of the synthetic conduit allows right-to-left shunting and depressurization of the lateral tunnel at the expense of systemic desaturation. C. Extracardiac Fontan operation is the current standard. The entire RA is bypassed by a synthetic conduit from the inferior vena cava directly to the RPA. The SVC is sutured directly to the RPA.
Ventricular arrhythmias may occur in a variety of settings, particularly after repair of ToF. The first generation of intracardiac repairs were performed via a large anterior ventriculotomy and frequently included incision of the pulmonary valve annulus and placement of a transannular patch made of pericardium or synthetic material (eg, Gore-Tex or Dacron). This technique successfully relieved the outflow tract obstruction but inevitably resulted in pulmonary valvar incompetence and pulmonary regurgitation (Fig. 84–9). Pulmonary regurgitation was thought to have minimal adverse clinical consequences, an assertion that held generally true for the first 2 decades after transannular patch repair. However, with time, the adverse hemodynamic effects of severe low-pressure pulmonary regurgitation become evident. The RV gradually dilates, becomes hypokinetic, and wall stress invariably increases. Ventricular tachycardia is likely to occur under such conditions and often arises from the region of the transannular patch or ventriculotomy suture lines. Programmed ventricular stimulation resulting in monomorphic or polymorphic ventricular tachycardia is of prognostic importance in these patients.69 In a cohort of 793 patients with repaired ToF followed for a mean 21.1 years, ventricular tachycardia occurred in 4.2% and sudden cardiac death occurred in 2% of patients.19 Transventricular and transannular repair were associated with ventricular tachycardia and sudden cardiac death. QRS width on the surface ECG has additive prognostic value. Gatzoulis and colleagues19 reported 88% of such patients with ventricular tachycardia and 63% of patients with sudden death had a QRS duration of 180 ms or above (see Fig. 84–15A). Patients invariably have a right bundle branch block that results from early injury because of surgical VSD closure and ventriculotomy followed by subsequent QRS lengthening secondary to RV dilatation. The presence of ventricular tachycardia (spontaneous or induced; monomorphic or polymorphic) in a patient with moderate or severe pulmonary regurgitation is an indication for surgical pulmonary valve replacement and ventricular scar excision. Transvenous radiofrequency ablation may be deferred in favor of surgical cryoablation in patients requiring surgical valve replacement. Transvenous radiofrequency ablation is feasible in patients with repaired ToF who have ventricular tachycardia originating from ventriculotomy or transannular patch scar sites and should be attempted in patients who do not require surgical valve or conduit replacement.70-72 Transvenous defibrillator placement may be considered in patients who continue to have inducible ventricular arrhythmias or spontaneous symptomatic ventricular arrhythmias despite radiofrequency ablation or surgical repair. There is a disparity between the high frequency of ventricular arrhythmia and the much lower incidence of sudden death in this population.73 Prophylactic antiarrhythmic therapy has not demonstrated efficacy in asymptomatic patients, and there is insufficient evidence to advocate prophylactic antiarrhythmic therapy in these patients. Identification of asymptomatic individuals who are at risk for ventricular tachycardia or sudden cardiac death remains a challenge. A very wide QRS (>180 ms) on the surface ECG is a marker for increased risk but has low predictive accuracy in individual cases.74 LV diastolic dysfunction and elevated LV filling pressure are predictors of ventricular arrhythmias and appropriate defibrillation in adults with repaired ToF.75,76 Further refinements in risk stratification are necessary and may involve electrophysiologic testing, exercise testing, evaluation of ventricular late potentials using signal averaged ECG, heart rate variability, and presence of repolarization abnormalities.77-79
Figure 84–9.
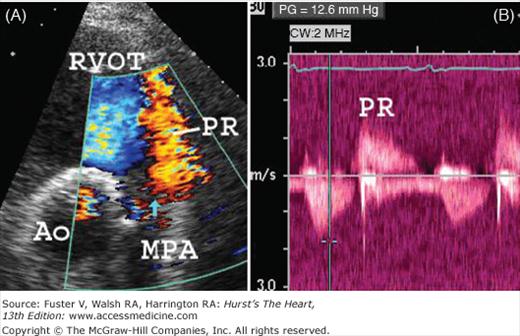
A 28-year-old patient with tetralogy of Fallot repaired during childhood with transannular patch placement. A. Transthoracic two-dimensional and color-flow Doppler in the parasternal short-axis projection demonstrating severe pulmonary regurgitation (PR) from the main pulmonary artery (MPA) to the right ventricular outflow tract (RVOT). The aortic valve is labeled (Ao). The blue vertical arrow points to a flail portion of the remaining pulmonary valve. B. Continuous-wave Doppler interrogation of the RVOT and MPA in the parasternal short-axis projection demonstrates a low-velocity (<2 m/s) jet of pulmonary regurgitation (PR) that ends before the onset of systole, consistent with elevated right ventricular diastolic pressure and restrictive right ventricular physiology. There is mild residual pulmonary stenosis.
An increasing number of women with complex CHD survive into adulthood because of the surgical advancements of the past 5 decades. This invariably means that an increasing number of women with complex CHD are reaching childbearing age, and many of them are reproducing. Pregnancy counseling is mandatory for all patients whether operated or unoperated. The maternal and fetal risks of pregnancy and transmitted risk of CHD should be discussed in women with CHD who are of childbearing age, preferably before pregnancy. Evaluation should include a detailed medical and social history with specific attention paid to functional capacity, which is an important prognostic determinant of maternal and fetal outcomes.80,81 Among risk factors for adverse outcomes, advanced New York Heart Association (NYHA) functional class and history of heart failure are well recognized. The detrimental effects of smoking include arrhythmias and increases in systemic vascular resistance, systemic blood pressure, pulmonary artery pressure and vascular resistance, atrial pressures, and heart rate.82,83 Transthoracic echocardiography determines ventricular and valvar function as well as pulmonary artery pressure. Impaired subpulmonary or subaortic ventricular function is associated with adverse maternal and fetal outcomes.80,81 Exercise echocardiography further assesses functional capacity, ventricular function, and valvar function.
The maternal cardiovascular system undergoes a myriad of changes during pregnancy that include a doubling of intravascular volume and cardiac output accompanied by a decrease in systemic vascular resistance. Obstructive lesions are less well tolerated than regurgitant lesions or left-to- right shunts. Severe aortic valve stenosis may be well tolerated if LV function is not impaired.81 When LV systolic function deteriorates, pregnancy becomes hazardous for the mother and the fetus. The maternal mortality rate for patients with a depressed ejection fraction and severe aortic stenosis is 5.9%.81 In general, women with valvular regurgitation and left-to-right shunts tolerate pregnancy well. However, patients with severe pulmonary regurgitation who have depressed subpulmonary ventricular function are at increased risk for adverse outcomes during pregnancy and at delivery.80 Heart failure followed by arrhythmia is the main complication of pregnancy in patients with various CHDs.
Patients with univentricular hearts or tricuspid atresia who have undergone the Fontan operation tolerate pregnancy well but have an increased risk of miscarriage.84 The outcomes may also be affected by single-ventricle morphology, specifically the decreased capacity of a morphologic RV to adapt to the volume and pressure changes that face the heart during pregnancy and the peripartum period. Thus, Fontan patients with an RV are at higher risk of developing congestive heart failure than those with an LV. Patients with repaired ToF, specifically those with depressed RV function and severe pulmonary regurgitation as a result of surgical valvotomy or transannular patch repair, are at an increased risk of adverse events.85 Thirteen percent of patients had cardiac complications in this study by Veldtman and colleagues.85 Decreased LV systolic function and pulmonary hypertension were similarly correlated with poor maternal and fetal outcomes. Six percent of infants in this study had CHD.
Patients with cyanosis face the most problems in carrying fetuses to term, and pregnancy is strongly advised against (see Table 84–2).86 There is a high incidence of early spontaneous abortion inversely related to the degree of cyanosis. In a study by Presbitero et al,86 only 12% of pregnancies in women with an oxygen saturation lower than 85% resulted in live births. The maternal complication rate has improved with close monitoring and meticulous care during pregnancy and delivery but is still considerable (32%). Maternal mortality in patients with Eisenmenger syndrome carries a 50% risk of mortality.87 There are scant data on the best way to manage patients seen late in pregnancy when safe termination is not feasible. Vaginal delivery is generally preferable because it results in less blood loss than cesarean section. Many maternal deaths occur after successful delivery when maternal intravascular volume and systemic vascular resistance increase, exceeding the capacity of the patient’s cardiovascular system to adapt. It is our practice at the Ahmanson/University of California, Los Angeles, Adult Congenital Heart Disease Center to advise our cyanotic patients against becoming pregnant and advise pregnancy termination if it is acceptable to the patient.
Patients with tissue abnormalities of the aorta, the worst extreme being Marfan syndrome, are at increased risk of aortic dissection or rupture if the ascending aorta is dilated (>40 mm).88,89 Patients with BAVs, independent of the degree of valvular stenosis, demonstrate a medial abnormality of the ascending aorta that places them at increased risk as well. Therapy with β-blockers in the peripartum period may decrease the risk of aortic dissection and rupture and is usually well tolerated. Severe valvular aortic stenosis places both the mother and fetus at risk because the decrease in systemic vascular resistance and large volume shifts during pregnancy and the postpartum period result in exaggerated valve gradients and increased filling pressure.90,91 Minimization of vascular volume and resistance alterations can be accomplished by avoidance of large IV volume infusions, avoidance of anesthetics known to decrease systemic vascular resistance, and use of adequate pain control to blunt increases in systemic blood pressure. Patients with severe aortic stenosis and congestive heart failure may require balloon valvuloplasty. The literature on aortic valvuloplasty in pregnancy consists of case reports and small case series. In our experience, balloon dilatation of a calcified aortic valve can tear the valve leaflets and cause severe poorly tolerated aortic regurgitation. Pregnant patients with heavily calcified valves and significant aortic regurgitation who are in congestive heart failure or are hemodynamically unstable may require surgical valve placement and cesarean delivery of the fetus.92
The management of pregnant women with mechanical valve prostheses is challenging for a number of reasons. Pregnant women are inherently hypercoagulable, and their risk of valve thrombosis is increased, thus necessitating appropriate anticoagulation.93,94 Oral warfarin accomplishes this task well, but it is teratogenic to fetuses, especially in the first trimester; the risk of teratogenicity is low after the eight week of gestation.95 Unfractionated heparin can be given subcutaneously twice a day or as a continuous IV infusion; even with meticulous control, there is still an increased risk of valve thrombosis.93,96 Subcutaneous low-molecular-weight heparin (LMWH) is more effective but requires frequent anti-Xa level monitoring.93 Some authors have advocated the use of low-dose warfarin throughout pregnancy because of the low risk of teratogenicity if the dose is less than 5 mg/d.94 Nonetheless, warfarin is generally avoided by most clinicians in favor of heparin and LMWH with careful partial thromboplastin time and anti-Xa levels, respectively.
The hemodynamic deteriorations during and after labor can be minimized by avoidance of rapid volume and pressure shifts. Planned induction 1 to 2 weeks before the expected term minimizes the risk of spontaneous labor and delivery. Invasive hemodynamic monitoring has not been shown to favorably affect outcomes and is often reserved for the highest risk subsets of patients (ie, those with critical valvular stenosis, severely depressed ventricular function with heart failure, or severe pulmonary hypertension). Vaginal delivery is recommended unless there are obstetric contraindications such as a fetus in the breach position. The use of antibiotics for endocarditis prophylaxis after rupture of the membranes is controversial, but most cardiologists recommend antibiotics under these circumstances for patients at high risk for developing infective endocarditis (see Table 84–1).
CHD is the leading cause of birth defects and noninfectious mortality in the first year of life.97 The cause of CHD is multifactorial. Genetic and environmental factors each play an important role in the development of CHD. Environmental insults such as chemical teratogens (eg, retinoic acid, lithium) and viral infections (eg, rubella) are known to increase the risk of CHD. Genetic factors also play a role in the development of CHD as demonstrated by the increased incidence of CHD in patients with chromosomal abnormalities such as microdeletions of chromosome 22q11 and trisomy 21. Most defects are not part of a syndrome, and patients have no family history of CHD. Even in seemingly sporadic cases of CHD, epidemiologic evidence demonstrates an increased likelihood of recurrence of CHD in subsequent pregnancies, supporting the existence of a genetic predisposition to CHD. It has been estimated that approximately 8% of CHD cases are caused by inherited genetic abnormalities.98 In an analysis of 6640 pregnancies in patients with a first-degree family history of CHD, the observed incidence of CHD in pregnancies referred because of sibling CHD and paternal CHD was 2% to 3%.99 There was a similar incidence of CHD for pregnancies referred because of maternal CHD (2.9%) or paternal CHD (2.2%). A study of a 1094 patients with CHD demonstrated an increase risk of CHD in the offspring of mothers with CHD (5.7%) versus fathers with CHD (2.2%).100 This study did challenge the polygenic basis for all forms of CHD by demonstrating that AV septal defect is a single-gene defect and ToF is a polygenic disorder with a small number of interacting genes. Isolated TGA is likely a sporadic defect. The risk of recurrence of CHD increases with the number of affected siblings or relatives, and genetic counseling should be provided for potential parents.101
The molecular mechanisms leading to CHD are complex, and the causes of the cardiac malformations observed in humans are still unclear. Furthermore, most patients do not have family history of CHD; thus, the mutations are likely spontaneous. The combining of expertise in cardiac anatomy, pathology, and molecular genetics is essential to adequately comprehend developmental national DNA bank of the CONCOR (CONgenital CORvitia) registry of CHD, will help facilitate research on the genetic contribution to CHD, and allow investigation into the molecular basis of CHD.102
Cardiac catheterization with angiocardiography has long been the “gold standard” for making and confirming hemodynamic and anatomic findings in patients with CHD. This position of preeminence is being challenged in the current era by an array of noninvasive imaging techniques, each with certain strengths and weaknesses, yet together capable of almost complete noninvasive anatomic and functional cardiovascular assessment. Transthoracic echocardiography with Doppler is the most widely used and cost-effective tool for diagnostic imaging in patients with CHD. Two-dimensional and more recently three-dimensional echocardiography provide a plethora of anatomic and functional data in a matter of minutes at low cost and without any radiation exposure (Fig. 84–10). Continuous-wave, pulsed-wave, and color-flow Doppler have added incremental value and are the most widely used techniques for quantifying valvar pathology and estimating intracardiac pressure. Tissue Doppler (a form of pulsed-wave Doppler) allows quantification of myocardial velocities and determination of ventricular electromechanical synchrony. The various Doppler-based methods may be used together to estimate atrial pressures, pulmonary artery pressure, and ventricular diastolic function.103 For example, the ratio of the early diastolic ventricular filling velocity (E) to the early diastolic mitral annular velocity (E’) provides an accurate estimate of atrial pressure.104 Tissue Doppler and speckle tracking for determination of ventricular strain and torsion provide a plethora of information regarding ventricular systolic and diastolic function.105
The Achilles heel of transthoracic echocardiography is the difficulty of image acquisition in certain patients such as obese patients and those with chest wall abnormalities as a result of previous surgeries.106 Moreover, posterior cardiac structures, such as pulmonary veins and atrial appendages, are often inadequately visualized. TEE traverses many of the above-mentioned hurdles of transthoracic echocardiography and is ideal for the visualization of pulmonary venous anatomy, atrial anatomy, AV valve morphology, ventricular outflow tract lesions, and vegetations or thrombi. Intraoperative TEE is particularly helpful during repair of congenital defects.
In parallel with the decreasing need for diagnostic catheterization, there has been a dramatic increase in the number of transcatheter cardiac interventions in patients with CHD.107 The majority of secundum ASDs can now be closed via transcatheter delivery of a closure device (Fig. 84–11). The success of percutaneous transcatheter closure of ASDs is contingent upon proper patient selection.108 TEE is widely used to assess the size, shape, and rim adequacy of ASDs and to exclude the presence of atrial thrombi or an anomalous pulmonary venous connection. Intracardiac echocardiography can also be used to guide ASD closure.
Figure 84–11.
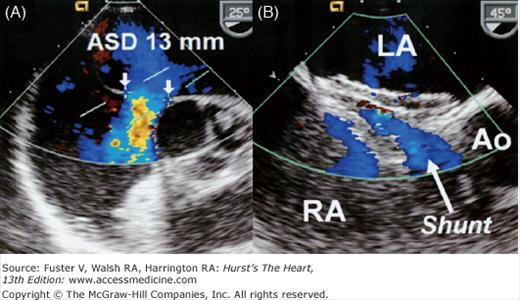
A. Transesophageal two-dimensional echo with color-flow Doppler demonstrating a 13-mm secundum atrial septal defect (ASD) with a thin posterior rim and an absent anterior (retro-aortic) rim. Note the left-to-right shunt (blue color-flow Doppler signal) across the ASD. B. The same patient after ASD closure of the ASD with an Amplatzer septal occluder. A small central left-to-right (arrow) from the left atrium (LA) to the right atrium (RA) is a common finding after device closure. The shunt usually resolves within a few weeks as the device endothelializes.
Cardiovascular magnetic resonance imaging (MRI) provides important incremental anatomic and functional data in patients with CHD. Ultrasound waves are not propagated through air; therefore, echocardiography (transthoracic or transesophageal) is of limited utility for imaging the extracardiac intrapulmonary vasculature, the site of frequent pathology in certain subsets of CHD. MR angiography (MRA) is invaluable in identifying the size, course, and degree of obstruction of medium to large vascular structures. Velocity mapping and flow quantification provide hemodynamic data (eg, the volume of pulmonary valve regurgitation per cardiac cycle).109 Gradient-echo cine-MRI provides incremental information regarding ventricular systolic and diastolic wall motion, segmental strain, and ventricular volumes (Fig. 84–12).110,111 Myocardial fibrosis detected by late gadolinium enhancement MRI is common in patients with repaired ToF.112 The degree of late gadolinium enhancement is related to adverse clinical markers, including ventricular dysfunction, exercise intolerance, neurohormonal activation, and arrhythmia. MRA of the coronary arteries is feasible, but imaging beyond the proximal coronary tree is suboptimal. Metallic structures, such as stents and valves, alter the local magnetic field and result in artifacts, thus limiting the utility of cardiac MRI in a significant subset of patients. The presence of a permanent pacemaker or defibrillator is considered a relative contraindication for cardiac MRI. The issues surrounding pacemakers are complex. Various generations of pulse generators and leads make generalizations regarding suitability for MRI problematic. Therefore, patients with pacemakers should not be scanned unless special circumstances arise and then only in centers with special cardiac MRI experience.
Figure 84–12.
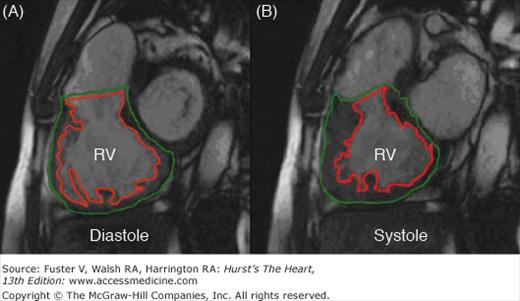
A stack of contiguous sagittal (short-axis) tomographic slices is acquired using cine-magnetic resonance imaging from the base to the apex of the heart. A. Short-axis image at the level of the mid right ventricle (RV) at end-diastole. The endocardial (red) and epicardial (green) borders are traced. B. The RV at end-systole with the endocardial and epicardial borders defined. These borders are defined in a stack of images from the base to the apex of the RV. According to Simpson’s rule, the ventricular volume is equal to the sum of the endocardial areas multiplied by the distance between the centers of each slice. The systolic and diastolic volumes obtained by this method are independent of geometric assumptions and dimensionally accurate.110
Cardiac CT has developed into a widely used noninvasive method for defining anatomy. ECG-gated multidetector CT provides tomographic two-dimensional data that can be sculpted into three-dimensional images, thus clarifying complex anatomy and enhancing the understanding of structure and form (Fig. 84–13).113 MRI tomographic images can similarly be reconstructed into three-dimensional structures. CT angiography (CTA) is gaining acclaim and experiencing exponential growth because of its capability to visualize the coronary arterial tree, with a high sensitivity, specificity, and negative predictive value for identifying obstructive coronary disease.114 CT has outperformed MRA in this respect. CTA has another advantage in that it does not require as much specialized training and technical knowledge as MRI.115,116 For these reasons, CTA is set to become the method of choice for noninvasive cardiovascular angiography in community practice while cardiac MRI remains confined to specialized regional centers despite certain advantages of MRI over CT (eg, the lack of radiation exposure and superiority in the provision of functional information functional). Moreover, cardiac CT can be performed in the presence of metallic structures or pacemakers, a relative contraindication for MRI.
Figure 84–13.

Computed tomography angiogram with three-dimensional surface rendering of a patient with a large secundum atrial septal defect (ASD) as viewed from a shallow right anterior oblique and slightly caudal perspective. Three-dimensional rendering software has been used to remove the anterior wall of the right atrium and right ventricle (RV). The inset demonstrates an outline of the ASD with division of the atrial septal rim into six quadrants. Assessment of three-dimensional ASD size and rim adequacy facilitates appropriate patient selection for percutaneous versus surgical defect closure.190 Ao, aorta.
Cardiac catheterization with angiocardiography remains the gold standard method for identifying anatomy, quantifying shunts, and measuring hemodynamics. Although the less invasive methods described above often obviate the need for invasive evaluation, numerous circumstances necessitate invasive evaluation. These include cases in which the noninvasive techniques give inconsistent or conflicting data or cases in which exact pressures and resistances must be measured. For example, patients with single-ventricle physiology may be candidates for the Fontan operation if certain hemodynamic and functional criteria are met (Table 84–3). Direct invasive measurement of pulmonary artery pressure and calculation of resistance must be performed to make the decision to proceed or not proceed with the Fontan operation.
| Morphologic criteria |
| Tricuspid atresia with pulmonary stenosis or atresia |
| Single ventricle with pulmonary stenosis or atresia |
| Hypoplastic ventricle that precludes biventricular repair |
| Hemodynamic criteria |
| Pulmonary arterial mean pressure <15 mm Hg |
| Pulmonary arterial mean pressure ⩽22 mm Hg if the Qp:Qs ratio ≥2:1 or aortic saturation reaches 85%, provided the pulmonary vascular resistance is <4-5 Wood units x m2 |
| Normal or mildly decreased ventricular systolic function (LV ejection fraction ≥50%; RV ejection fraction ≥40%) |
| No more than mild valve regurgitation |
| Ventricular end-diastolic pressure ≤12 mm Hg |
Exercise and pharmacologic stress testing provide prognostically important information regarding maximum exercise capacity, cardiopulmonary reserve, and chronotropic capacity.25,117 Exercise capacity is generally depressed in adults with CHD, and drastically so in patients with Eisenmenger syndrome (see Fig. 84–4). The decreased exercise capacity in patients with CHD is comparable to patients with ischemic and nonischemic cardiomyopathy. Chronotropic incompetence is common in certain subsets of patients, specifically those with previous operations or taking β-blockers. Lack of a heart rate response to exercise, pulmonary arterial hypertension, and impaired ventilatory function are correlates of maximum exercise capacity. A blunted heart rate response to exercise identifies patients at increased risk of dying.118 Cardiopulmonary exercise testing adds incremental diagnostic and prognostic information in patients with CHD. Maximum oxygen consumption (VO2max) is decreased in patients with CHD (see Fig. 84–4). Poor exercise capacity identifies adult CHD patients at risk for hospitalization or death. Cyanotic patients with right-to-left shunts (eg, Eisenmenger syndrome) fail to demonstrate a normal increase in pulmonary blood flow with exercise. The systemic arterial resistance decreases with no appreciable change in pulmonary arterial resistance; therefore, more deoxygenated blood is shunted away from the pulmonary arterial bed, and patients desaturate with exercise. The carotid chemoreceptors located at the bifurcation of the internal and external carotid arteries detect this hypoxia, and via a complex feedback mechanism involving the respiratory control centers of the brain stem, increase the minute ventilation, resulting in hyperventilation during exercise (Table 84–4). Ventilatory response to exercise is abnormal across the spectrum of CHD but is most markedly abnormal in cyanotic patients irrespective of pulmonary arterial hypertension. An increased ratio of minute ventilation to CO2 production (VE/VCO2) slope is a strong exercise predictor of death in noncyanotic CHD patients.26 Oxygen uptake (VO2) slowly increases in the later stages of exercise yet remains markedly reduced in patients with cyanotic heart disease (see Fig. 84–4). Submaximal exercise capacity as measured by 6-minute walk distance is used widely and is of prognostic importance in patients with pulmonary arterial hypertension and correlates closely with peak VO2.119
| Low peak VO2 |
| Low AT |
| Reduced phase I VO2 |
| Increased ventilatory response to exercise (VE/VCO2) |
| Immediate worsening of hypoxemia at start of exercise |
Figure 84–14.
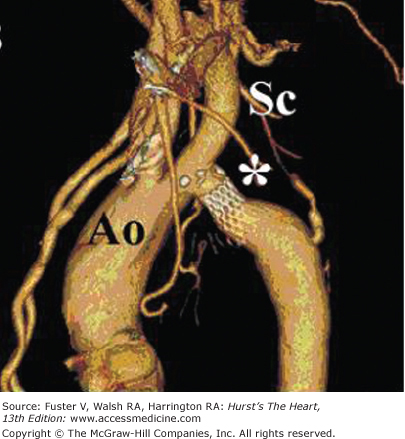
Electrocardiography-gated cardiovascular CT angiogram with three-dimensional reconstruction viewed from the left anterior oblique projection in a 46-year-old patient with a native coarctation of the aorta (Ao) after transcatheter stent implantation (asterisk). The stent appears widely patent and does not intrude upon the ostium of the left subclavian artery (Sc).
Psychosocial Aspects
As patients with CHD make the transition from adolescence to adulthood, it is imperative that they come to understand the nature and implications of their heart problem and what interventions have and may need to be performed. Appropriate advice and guidance should be available regarding employment, insurance, socialization, contraception, and exercise.
The majority of operated and unoperated patients can be and are gainfully employed. Patients with simple forms of CHD, such as isolated VSD, pulmonary stenosis, or aortic valve stenosis, often achieve higher levels of education then the national standards.120 A cross-sectional study from Holland in which CHD patients were asked to complete a questionnaire focusing on employment issues demonstrated that adults with complex CHD have reduced job participation compared with patients with mild CHD (59% vs 76% employed).121 Many receive disability benefits or experience career problems or job handicaps. Career counseling focusing on physical abilities and level of education may help increase appropriate employment in this population. Adults with CHD are at a disadvantage when applying for employment because of the tendency of some employers to consider the potential for future deterioration of an applicant with a “known heart problem.” In the United States, the National Rehabilitation Act of 1973 obliges employers to consider only the present capacity of a prospective employee to perform a given job.
Restrictions for employment do exist for certain jobs in which the safety of others is the direct responsibility of the patient with CHD. The clearest example of this is commercial airline pilots. It is the task of CHD specialists to estimate the risk of acute disability or sudden cardiac death in such patients. Low-risk rates are defined in only a small subset of patients with CHD.122
Adults with CHD are significantly more likely to have difficulty obtaining life insurance.123 Refusal rates appear to be independent of the type or severity of CHD, suggesting that the label of CHD has a negative impact by itself. This problem is compounded by the limited long-term survival data for specific CHD subtypes. An article by Vonder Muhll et al124 discuss the scope of this problem and provide a summary of mortality data and predictors of adverse outcomes in various subsets of CHD along with practical tips for patients and physicians to assist with the insurance application process.124 In general, insurance policies are restrictive, and patients with CHD are frequently insured at higher rates or not insured at all.125
As a result, the patient must seek to obtain a new policy, which often excludes benefits for medical or surgical treatment of the congenital cardiovascular condition. In 2010, a health care bill was passed by the Congress of the United States which included extension of parental coverage until 26 years of age. Despite a call for near universal health care coverage, it remains unclear how such an initiative will be funded at the federal and state level.
The transition from adolescence to adulthood is difficult enough for many patients with CHD and is often made more difficult by the cessation of health insurance under a parent’s policy. As a result, the patient must seek to obtain a new policy, which often excludes benefits for medical or surgical treatment of the congenital cardiovascular condition. In a pilot study by Moons et al126 investigating the expenditure on health care of adults with CHD, patients with CHD incurred considerably higher expenses than age- and gender-matched control subjects. This difference, however, was accounted for by a minority of patients with CHD, specifically those with dilated LVs, decreased functional capacity, women, and older patients. Further data are clearly needed to predict the potential health care expenditure in various subsets of patients with CHD.
Most adults with CHD appear well adjusted. Utens et al127 assessed the occurrence of a wide range of behavioral and emotional problems in 166 young adults long term after surgical correction compared with age-matched control subjects using the Young Adult Self-Report.127 On most Young Adult Self-Report scales, no differences were found between the mean problem scores of the CHD patients and control subjects except for small differences in two scales (Somatic Complaints and Strange). No significant relationship was found between cardiac diagnosis and problem behaviors in CHD adults. Moreover, no relationship was found between IQ scores and problem behaviors. Psychosocial problems can occur in patients with CHD, manifesting in excessive psychologic stress that is unrelated to the clinical severity of the original cardiac defect.128 In a study by Ternestedt et al,129 two cohorts of patients operated on before the age of 15 years, one for ToF and the other for ASD, were queried 20 and 30 years after surgical repair regarding quality of life. There was no connection between quality of life and functional capacity. Interestingly, fewer patients in the ToF than in the ASD group considered that their lives were affected by their heart disease. Therefore, multiple questionnaire-based studies have demonstrated that the severity of the heart disease is not necessarily congruent with estimated quality of life, suggesting that patients develop coping strategies during childhood and adolescence.
Although most patients with CHD appear well adjusted, many have the feeling that they are different from their peers and suffer experience isolation and low self-esteem, feelings compounded by exercise limitations, surgical scars, and parental overprotection. These factors, when added to the challenges of adolescence and young adulthood, can adversely affect psychosocial development. A number of large longitudinal series of patients with repaired ToF have demonstrated that suicide is a common cause of late death, ranging in frequency from 4.8% to 10%.130-132 Psychosocial anxiety and neurotic behavior may be avoided by early repair in childhood.133 Anxieties about sexuality, relationships, and reproduction are common and often prove difficult to adequately discuss with the physician in a busy clinic setting; such issues are more thoroughly dealt with by a multispecialty team that may include a psychologist, social worker, and nurse practitioner. Psychological support should be considered as one of the most important aspects in the long-term care of patients with CHD.
The impact of CHD on intellectual development appears more dependent on lesion type and severity than psychosocial issues are. Studies of intellectual development and capacity mostly exclude patients with genetic syndromes or overt neurologic defects. Cyanosis is associated with mild intellectual impairment; however, it is unclear that early repair of cyanotic lesions such as ToF results in improved intellectual function.134,135 Neurodevelopmental outcomes in patients with single-ventricle physiology who have undergone the Fontan operation are in the normal range, but performance on examinations of IQ are lower than the general population.136



