Congenital Heart Disease
M. Elizabeth Brickner
Introduction
Congenital heart disease (CHD) is defined as a gross structural abnormality of the heart, great arteries, or great veins that is present at birth (1). Congenital cardiac malformations are relatively uncommon. Multiple studies have demonstrated an incidence of 0.6% of live births for moderate to severe defects (2). The prevalence is higher in stillbirths and spontaneous abortions (3). There are approximately 32,000 new cases per year in the United States and greater than 1,000,000 new cases per year worldwide (4). Although the prevalence is low, the population of patients with CHD continues to expand. Because of the dramatic advances made in medical, surgical, and interventional device therapy, survival into adulthood is now the rule for the vast majority of patients with congenital cardiac defects.
Etiology
The etiology of CHD is multifactorial. Recurrence risks vary with the gender of the proband and the specific cardiac defect (5), with an overall recurrence risk of 3% to 5% in the offspring of patients with congenital heart disease (6,7,8). The exact proportion of patients with a specific genetic etiology is unknown. There are reports of familial defects following Mendelian patterns of inheritance (9). Certain chromosomal abnormalities are associated with congenital heart defects. The most common of these is trisomy 21 (Down syndrome). At least 50% of patients with Down syndrome have CHD (most commonly atrioventricular septal defects or ventricular septal defects), often associated with early pulmonary vascular obstructive disease (1,11). Despite the identification of many candidate genes on chromosome 21, the key genes contributing to the cardiac phenotype in Down syndrome have yet to be defined (12). Other syndromes associated with CHD include Turner syndrome, Noonan syndrome, Williams syndrome, Marfan syndrome, and trisomy 13, 14, 15, and 18.
Deletions of 22q11 are seen in DiGeorge syndrome (thymic aplasia, hypoparathyroidism, congenital heart defects involving the outflow tracts, and a dysmorphic appearance) (13,14). From transgenic studies with a mouse model, TBX1 has been identified as the likely gene responsible for the cardiac and hypoparathyroid phenotype (15). 22q11 deletions are now recognized as the cause of a broader group of defects and are seen in 50% of patients with conotruncal abnormalities (14). CATCH-22, a syndrome due to microdeletion at chromosome 22q11, consists of cardiac conotruncal abnormalities, abnormal facies, thymic abnormalities, cleft palate, and hypocalcemia. Some patients may have the gene deletion without accompanying syndromic features (13).
Mutations in a few specific genes have been identified in some cases of congenital heart defects. Mutations in TBX5 are seen in the majority of patients with Holt-Oram syndrome, an autosomal disorder with cardiac septal defects and upper limb defects (16). Mutations in the elastin gene (ELN) have been identified as a cause of supravalvular aortic stenosis (17). Mutations in NKX2.5 have been associated with the autosomal dominant phenotype of atrial septal defect or tetralogy of Fallot (18).
Fetal and Neonatal Circulation
In fetal life, the placenta is a low-resistance structure that acts as a respiratory organ and receives the largest amount of fetal blood flow. Blood from the placenta returns to the fetus through the ductus venosus, entering the inferior vena cava (IVC) to the right atrium (RA). A portion of the IVC blood flow is directed across the patent foramen ovale (PFO) to the left atrium (LA), bypassing the right heart. Blood from the superior vena cava (SVC) is directed into the right ventricle (RV) along with the remaining blood return from the IVC and is then pumped out into the pulmonary artery. Because of high pulmonary vascular resistance in the fetus, most pulmonary blood flow crosses the
ductus arteriosus and enters the descending thoracic aorta. At birth, the relatively low resistance placental circulation is removed, and systemic vascular resistance increases within minutes. With respiration, the pulmonary vascular bed dilates in response to inspired oxygen, and pulmonary vascular resistance decreases while pulmonary blood flow increases. Pulmonary venous blood return increases, which increases systemic ventricular output and helps to close the foramen ovale. The ductus arteriosus is patent at birth, but begins to constrict shortly after birth and usually closes within hours to several days (1). Defects in which pulmonary blood flow depends on flow through the ductus are characterized as ductal dependent. With closure of the ductus in these patients, progressive hypoxemia, acidosis, and death invariably occur. Prostaglandin E1 infusion to maintain patency of the ductus is used as a temporizing measure until more definitive therapy can be undertaken (19).
ductus arteriosus and enters the descending thoracic aorta. At birth, the relatively low resistance placental circulation is removed, and systemic vascular resistance increases within minutes. With respiration, the pulmonary vascular bed dilates in response to inspired oxygen, and pulmonary vascular resistance decreases while pulmonary blood flow increases. Pulmonary venous blood return increases, which increases systemic ventricular output and helps to close the foramen ovale. The ductus arteriosus is patent at birth, but begins to constrict shortly after birth and usually closes within hours to several days (1). Defects in which pulmonary blood flow depends on flow through the ductus are characterized as ductal dependent. With closure of the ductus in these patients, progressive hypoxemia, acidosis, and death invariably occur. Prostaglandin E1 infusion to maintain patency of the ductus is used as a temporizing measure until more definitive therapy can be undertaken (19).
Diagnostic Tools
The physical exam is critical in the evaluation of patients with known or suspected CHD and includes elements that may not be routinely performed in patients with acquired forms of heart disease. In addition to precordial palpation, assessment of venous waveforms, and careful cardiac auscultation, it is also important to assess for cyanosis (including differential cyanosis), palpate pulses and measure blood pressure in both upper and lower extremities, and check oxygen saturation. Evidence of phenotypes associated with CHD (e.g., Down syndrome, William syndrome) should be sought. Although the electrocardiogram (ECG) and chest x-ray (CXR) are a routine part of the evaluation of patients with CHD, they are not specific enough for diagnostic purposes. Imaging studies by qualified personnel play a critical role in the evaluation of these patients. A careful review of prior data, including catheterization data, imaging, and operative reports, is essential as well.
Transthoracic echocardiography (TTE) is the most widely used diagnostic tool for establishing the initial diagnosis and following patients serially (20,21). Studies in these patients are complex and time-consuming and should be performed by sonographers and physicians with expertise in CHD. Transesophageal echocardiography (TEE) is particularly useful in adults with poor acoustic windows, providing excellent visualization of the atrial septum, pulmonary veins, interatrial baffles, and Fontan connections (22,23). Intraoperative TEE plays a critical role for patients undergoing surgical repair (24). TEE is also used to guide catheter interventions and device deployment. Increasingly, intracardiac echocardiography is being used for these procedures. Three-dimensional echocardiography is an evolving technology that may be helpful in evaluating patients with congenital heart disease (25,26).
Cardiac magnetic resonance imaging (MRI) is an extremely useful tool for the assessment of patients with congenital heart disease, providing high-quality images with a wide field of view in nearly all patients. MRI is particularly useful for assessment of extracardiac anatomy, including delineation of the great vessels, branch pulmonary arteries, and surgical shunts, as well as systemic and pulmonary venous connections (27,28,29). MRI allows quantitation of ventricular mass, volumes, and ejection fraction and can be used to calculate shunt flow and regurgitant flow. Contrast is not required for routine imaging but may be particularly useful in assessing vascular structures. The role of cardiac computed tomography (CT) imaging is evolving. Cardiac CT provides excellent visualization of anatomy (particularly extracardiac anatomy) but does use ionizing radiation (30).
Cardiac catheterization plays a critical role in the management of patients with congenital heart disease, as both a diagnostic and a therapeutic tool (31,32). Due to the complex hemodynamic data and difficult anatomy in many of these patients, catheterization is best performed by experienced personnel. There is an expanding role for interventional catheterization procedures, including closure of shunts, pulmonary and aortic valvotomy, pulmonary artery stenting, stenting of conduits, and balloon aortoplasty for aortic coarctation. (See Chapter 83.)
Specific Defects
Left-to-Right Shunts
Atrial Septal Defect
An atrial septal defect (ASD) is a direct communication between the atrial chambers. ASDs are common, accounting for 5% to 10% of all congenital heart defects and one third of all congenital defects diagnosed in adulthood (33,34). They are usually sporadic, but familial cases have been reported. They are more common in female than male individuals (2:1) (35). An associated congenital defect may be seen in up to 30% of cases. ASDs are seen in association with skeletal deformities of the upper extremities, including Holt-Oram syndrome (36). Ostium secundum and primum defects are also associated with Down syndrome.
There are several morphologic types of ASDs. The most common is the ostium secundum defect (seen in 75% of cases). Secundum defect occurs in the region of the fossa ovalis, may extend in any direction, and may be multiple. Partial anomalous pulmonary venous connections are seen in 2% of patients with secundum defects. Ostium primum defects account for 15% of ASDs. Primum defects are part of the spectrum of atrioventricular (AV) septal defects and are associated with a common AV junction. A common AV valve is usually present with fusion of the inferior and superior bridging leaflets, leading to separate mitral and tricuspid orifices. This results in a trileaflet appearance of the “anterior” mitral leaflet, sometimes referred to as “cleft” in the anterior leaflet. Mitral regurgitation may be associated with this abnormal valve. Sinus venosus defects account for 10% of ASDs. Sinus venosus defects occur in the superior portion of the septum near the insertion of the SVC and are frequently associated with anomalous pulmonary venous drainage of the right pulmonary veins, most commonly the right upper pulmonary vein. Inferior sinus venosus defects are rare. They occur at the mouth of the IVC and may have right-to-left shunting and cyanosis due to preferential shunting of IVC blood to the LA. The rarest form of ASD is the coronary sinus defect, which may occur at the mouth of the coronary sinus or in the body of the coronary sinus itself (known as “unroofing” of the coronary sinus). Coronary sinus ASDs are often associated with a persistent left superior vena cava connecting to the LA (37). Some patients may have absence of most of the interatrial septum, resulting in a “common atrium.”
With unrestricted defects, there is no pressure gradient between the atria. Left-to-right shunting across the ASD occurs in late systole and diastole. The magnitude of the shunt depends on the size of the defect and the relative of compliance of the right and left ventricles as well as the pulmonary and systemic vascular resistance. Diseases that affect left ventricular (LV) compliance (e.g., hypertension, coronary artery disease) can increase the magnitude of the left-to-right shunt. The left-to-right shunt results in right ventricular volume overload with increased pulmonary blood flow (Fig. 30.1). Large shunts may result in pulmonary hypertension. Spontaneous closure of ASDs may occur (38,39,40). Small ASDs (<3 mm) usually close by 18 months, and as many as 80% of defects in the range
of 5 to 8 mm close by 18 months. Larger defects rarely close spontaneously.
of 5 to 8 mm close by 18 months. Larger defects rarely close spontaneously.
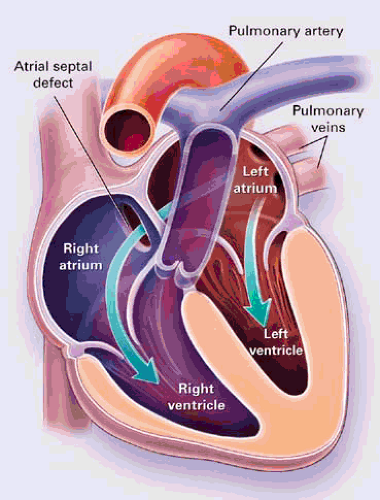 FIGURE 30.1. Left-to-right shunting across an ostium secundum atrial septal defect, causing right ventricular volume overload. (From Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. N Engl J Med 2000;342:340. ) |
The cardiac exam demonstrates a right ventricular lift with significant volume overload. S1 is normal. S2 is widely split and does not vary with respiration, although this pathognomonic finding is not universally present. A systolic flow murmur is common due to increased flow across the right ventricular outflow tract. A diastolic rumble across the tricuspid valve may be heard with large shunts. With the development of pulmonary hypertension, splitting of S2 narrows and the intensity of P2 increases. With shunt reversal (the Eisenmenger syndrome), cyanosis and clubbing develop. Cyanosis may also be seen in the absence of pulmonary hypertension in patients with very large defects, a prominent Eustachian valve, a coronary sinus defect, or in association with pulmonic stenosis, RV dysfunction, or Ebstein’s anomaly. Typical ECG findings include right-axis deviation (except in ostium primum defects) and an rSR′′ or rsR′ pattern in lead V1. There may be evidence of right ventricular hypertrophy (RVH). Some patients have prolongation of the PR interval. Inverted P waves in the inferior leads suggest a sinus venosus type of defect. A superior QRS axis (extreme right- or left-axis deviation) suggests a primum atrial septal defect. The CXR shows right-sided chamber enlargement, a dilated pulmonary artery, and increased pulmonary vascular markings in patients with significant shunts.
The diagnosis is made by echocardiography, which demonstrates the location and size of the defect as well as the direction of shunting. The presence of a dilated RA and RV consistent with right-sided volume overload should suggest the presence of an ASD, prompting thorough interrogation of the atrial septum and a bubble study. Ostium secundum and primum defects are well visualized by transthoracic imaging, particularly on subcostal views. Sinus venosus defects may be more difficult to demonstrate and require additional views (41,42). TEE is frequently used in the adult populations to fully interrogate the interatrial septum (43).
Cardiac catheterization is not usually required in patients with an ASD unless there is associated pulmonary hypertension or the noninvasive assessment is inconclusive (44). The presence of an ASD is confirmed by catheter passage across the atrial septum and a “step up” in oxygen saturation at the level of the atrium. Systemic and pulmonary blood flow, ratio of pulmonary to systemic blood flow (Qp/Qs), pulmonary pressure, and pulmonary vascular resistance should be assessed. If anomalous pulmonary venous drainage is suspected, levophase pulmonary artery injections should be obtained. Coronary angiography is usually performed for patients over the age of 40 years if surgical correction is planned. ASDs can also be diagnosed by cardiac MRI, which is also excellent for assessing pulmonary venous connections.
ASDs are often diagnosed in childhood, but they can also present in adulthood. Patients with an ASD are usually asymptomatic in childhood, but patients may have decreased exercise tolerance and increased respiratory infections. Symptoms usually occur in adulthood by the third or fourth decade. Seventy percent of patients will have symptoms by the fifth decade, and annual mortality increases to 10% by the sixth decade for patients with untreated ASDs (45). The most common symptoms are dyspnea and decreased exercise tolerance. Patients may present with atrial arrhythmias, congestive heart failure, or symptoms associated with pulmonary vascular disease. There is some increased risk of stroke due to paradoxical embolism, but this is usually seen only in patients with atrial arrhythmias and/or right ventricular dysfunction. Pulmonary vascular obstructive disease (the Eisenmenger syndrome) is uncommon with ASDs, occurring in 5% to 10% of cases, more commonly in female patients (45,46). Patients with the Eisenmenger syndrome secondary to an ASD typically present in their twenties or thirties.
Closure of the defect is recommended for ASDs with a Qp/Qs greater than 1.5:1 and a pulmonary to systemic vascular resistance ratio less than 0.7 (47,48,49,50). In children, closure is usually recommended between the ages of 2 and 4 years to allow for spontaneous closure. In adolescents and adults, closure is usually undertaken when the diagnosis is made. The surgical approach has low morbidity and mortality (<1%) and is done by patching the defect or with direct suture closure.
Surgical closure of an ASD in childhood or early adulthood (before the age of 25 years) results in a long-term mortality similar to that of an age- and sex-matched control population (47). These patients can be considered cured. Patients undergoing surgery after the age of 25 years have reduced survival compared to control subjects, most strikingly in those older than the age of 40 years. Surgical closure between the ages of 25 and 40 years in asymptomatic patients is controversial but is generally presumed to prevent symptomatic deterioration (49,51,52). For symptomatic patients older than the age of 40 years, surgical closure improves exercise capacity, improves survival compared to medically managed patients, and prevents further deterioration in functional capacity (47,53). However, it does not reduce the risk of supraventricular arrhythmias, heart failure, or cerebrovascular accidents (48,53,54,55,56,57). Surgical closure in symptomatic patients with a significant shunt who are over the age of 60 years results in symptomatic improvement in 80% (54). Patients with older age at repair require surveillance for atrial arrhythmias, heart failure, stroke, and progressive pulmonary vascular disease. Seventy percent of patients with preoperative arrhythmias have persistent arrhythmias postoperatively, and 10% to 25% of patients without arrhythmias will develop them postoperatively. An increased risk of systemic arterial hypertension of unclear
etiology has been demonstrated after ASD closure in older patients (54).
etiology has been demonstrated after ASD closure in older patients (54).
Preoperative pulmonary vascular resistance (PVR) is predictive of outcome. Patients with a PVR of less than 7 Wood units have improvement in symptoms and New York Heart Association (NYHA) functional class, whereas a PVR of greater than 15 Wood units is associated with a high surgical mortality (50). If PVR is greater than two thirds of systemic vascular resistance, patients must have a large shunt or evidence of pulmonary vascular reactivity before surgery is considered.
The role of device closure is increasing (56). First attempted in 1976, device systems (57) have undergone continuous evolution in terms of material, design, shape, and delivery method. Devices available or in clinical trials include the Amplatzer septal occluder, the Atrial Septal Defect Occlusion System (ASDOS), the Buttoned Device, the Guardian Angel, the Helex Septal occluder, and the Cardioseal. Success rates vary but generally are in the range of 90% to 97% for initial successful deployment. Residual leaks are common on initial assessment but usually decrease or disappear with longer-term follow-up. Choice of the device type depends on the location of the defect and the degree of aortic rim tissue (must have 4- to 5-mm rim). There are no comparative studies between device and surgical closure. (See Chapter 83.)
There is no consensus for long-term follow-up after device closure, and long-term outcomes are largely unknown (including the risk for atrial arrhythmias, heart failure, and stroke). Because there is ongoing morbidity and mortality in adults undergoing surgical closure of ASDs, it is reasonable to postulate that a similar outcome may be seen in adults undergoing device closure. Potential complications specific to device closure include the potential for obstruction of pulmonary or systemic venous drainage, interference with the mitral valve, and erosion of the atrial wall or the aortic wall.
Ventricular Septal Defect
Ventricular septal defects (VSDs) are the most common form of congenital heart defect, accounting for 25% to 30% of all patients with congenital heart disease (33). The male:female ratio is 1. VSDs are the most common defect seen in the pediatric population. VSDs are usually a single defect, but they can occur in the setting of more complex congenital heart defects. Defects can be divided into restrictive defects (flow restricted between the LV and the RV with right ventricular pressure less than half of systemic levels) or nonrestrictive defects (with equal left and right ventricular pressures) (58,59). From 70% to 80% of VSDs can be characterized as restrictive, with the potential to close or become smaller (60,61). Nearly half of all VSDs are small, and up to 75% may close spontaneously. Even large defects can decrease in size (62,63,64,65). VSDs usually close by the age of 10 years. Spontaneous closure in adults is rare but has been reported (65).
The ventricular septum consists of the trabecular muscular septum, the inlet septum (formed from the endocardial cushion), the outlet or infundibular septum, and the membranous septum. Failure of growth, alignment, or fusion of these components results in a VSD. Perimembranous VSDs are the most common type, accounting for 75% to 80% of cases (Fig. 30.2). A perimembranous defect occurs at the junction of the inlet, outlet, and trabecular septum and may extend variably into these regions. The perimembranous VSD underlies the septal leaflet of the tricuspid valve and may decrease in size or close spontaneously due to adherence of septal leaflet tissue to the defect, resulting in a ventricular septal aneurysm. Inlet septal defects account for 5% to 10% of VSDs. They occur in the muscular septum, under the mitral and tricuspid leaflets, due to deficiency of tissue from the endocardial cushion. Inlet VSDs rarely close spontaneously. Muscular defects or defects of the trabecular septum account for 20% of all VSDs. They may be located in various positions within the trabecular septum and may be multiple. Muscular VSDs tend to decrease in size with muscle growth and may close spontaneously. Outlet defects (also known as doubly committed subarterial defects or supracristal VSDs) account for 5% of all VSDs. They occur in the right ventricular outlet or conal portion of the septum, underlying both the pulmonary and aortic valves. Outlet defects do not close spontaneously, but their size can decrease due to prolapse of aortic cusp tissue through the defect (also resulting in aortic regurgitation).
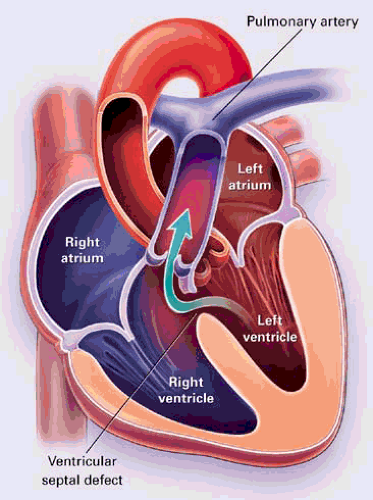 FIGURE 30.2. Left-to-right shunting across a perimembranous ventricular septal defect. (From Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. N Engl J Med 2000;342:340. ) |
The degree of left-to-right shunting depends on the size of the defect and the relative resistance of the systemic and pulmonary vascular beds. VSDs are characterized as small when the defect size is less than one-third of the aortic root size, and these are always restrictive. Pulmonary vascular resistance remains normal. With moderate restrictive defects, the defect is approximately half the size of the aortic valve, and there is moderate to severe left-to-right shunting. Patients with moderate defects may develop symptoms associated with LV volume overload and are at risk for developing pulmonary vascular disease. Large VSDs are nonrestrictive, with equal pressures in the left and right ventricles. There is a large left-to-right shunt initially, and the pulmonary circulation is exposed to systemic pressures early in the course of the disease. Patients with nonrestrictive VSDs usually develop irreversible pulmonary vascular disease within the first decade of life, eventually resulting in shunt reversal and Eisenmenger physiology.
The natural history of VSDs depends on the size and location of the defect. Small, restrictive defects with a Qp/Qs less than 1.5 to 1 do not place a hemodynamically significant load on the LV. Moderate or large defects cause pulmonary
congestion and LV volume overload, which may lead to LV dysfunction and congestive heart failure. Pulmonary hypertension may occur with moderate defects. Larger defects are associated with a significant risk of pulmonary hypertension and pulmonary vascular obstructive disease. The Eisenmenger syndrome occurs in 10% of patients with VSDs (65). All patients are at risk for bacterial endocarditis and require antibiotic prophylaxis. Other complications include aortic cusp prolapse through the defect, resulting in aortic regurgitation and/or subaortic obstruction, and the development of a double-chambered RV due to hypertrophy of muscle bundles within the mid-right ventricular cavity.
congestion and LV volume overload, which may lead to LV dysfunction and congestive heart failure. Pulmonary hypertension may occur with moderate defects. Larger defects are associated with a significant risk of pulmonary hypertension and pulmonary vascular obstructive disease. The Eisenmenger syndrome occurs in 10% of patients with VSDs (65). All patients are at risk for bacterial endocarditis and require antibiotic prophylaxis. Other complications include aortic cusp prolapse through the defect, resulting in aortic regurgitation and/or subaortic obstruction, and the development of a double-chambered RV due to hypertrophy of muscle bundles within the mid-right ventricular cavity.
The physical exam findings vary with the size of the defect. A patient with a small defect has a normal PMI, a normal S1 and S2, and a harsh pansystolic murmur associated with a systolic thrill. In addition to the murmur and thrill, patients with larger defects have evidence of LV enlargement with prominence and/or displacement of the apical impulse, a diastolic mitral inflow rumble, and frequently a gallop rhythm. With the development of pulmonary hypertension, the intensity of P2 increases, splitting of the second heart sound becomes narrowed, and the murmur decreases or disappears.
ECG findings are nonspecific. The ECG is normal with small defects. Larger defects are usually associated with the development of left ventricular hypertrophy (LVH) and ST-T wave changes. RVH may be seen with large defects or with the Eisenmenger syndrome. The CXR is normal with small defects, but cardiomegaly and pulmonary plethora are seen with larger defects. Patients with severe pulmonary vascular disease and shunt reversal (Eisenmenger physiology) have mild cardiomegaly or normal heart size with large central pulmonary arteries, peripheral pruning of the pulmonary vessels, and oligemic lung fields.
The diagnosis can be made by echocardiography with Doppler color flow mapping. With careful interrogation of the septum, the site and size of defects can be demonstrated (66,67,68). The pressure gradient between the LV and the RV can be assessed by continuous-wave Doppler interrogation of the VSD jet, and right ventricular systolic pressure can be indirectly estimated from continuous-wave Doppler interrogation of the tricuspid regurgitation (TR) jet (69,70). Care must be taken with the latter approach because the TR jet may be contaminated by the VSD jet (particularly with perimembranous defects), resulting in inaccurate right ventricular pressure estimation. The interventricular pressure gradient may be inaccurate in the setting of tortuous or serpiginous defects where the modified Bernoulli equation is not applicable. Echocardiography may also reveal other associated defects, including aortic regurgitation.
Cardiac catheterization is generally reserved for patients in whom there is uncertainty regarding the size of the shunt and the pulmonary vascular resistance. The reversibility of pulmonary hypertension can be assessed with the administration of oxygen, nitric oxide, prostaglandins, or adenosine. Selective coronary angiography is usually performed for patients older than the age of 40 years if surgical repair is planned.
The clinical presentation depends on the size of the shunt. Patients with small defects are asymptomatic and have normal growth and development. The diagnosis in usually made on the basis of finding a loud holosystolic murmur. Larger shunts may result in symptoms of congestive heart failure in infancy as well as an increased susceptibility to pulmonary infections. The diagnosis of a VSD in adulthood is usually based on the incidental finding of a murmur or the development of a complication related to the VSD (e.g., endocarditis, aortic valve prolapse and regurgitation, or the Eisenmenger syndrome) (71). Overall, the 25-year survival for all patients is 87% (65). Mortality increases with the size of the VSD.
Patients with symptomatic heart failure initially are treated with medical therapy, including diuretics and afterload reduction. Digoxin is often used in the pediatric setting. There are no randomized trials of medical therapy, but its use is indicated to stabilize the patient until surgical repair can be performed. Indications for surgery include severe intractable heart failure within the first 3 months of life, the presence of symptoms in older infants and children, and the presence of a moderate or large defect with a Qp/Qs greater than 2:1. Repair is also recommended for subarterial defects regardless of the shunt size due to the risk of aortic valve prolapse. Pulmonary vascular resistance should be below 8 Wood units (less than two-thirds systemic vascular resistance) for surgery to have long-term success. Repair is usually performed from the RA but occasionally through the RV, with placement of a patch or direct suture closure. Pulmonary artery banding is rarely performed. It is used to decrease pulmonary blood flow in patients with multiple defects or complex malformations that are not otherwise amenable to repair. Transcatheter device closure of VSDs appears to be feasible in some cases but is not widely available (72,73,74,75).
The prognosis is normal for patients with spontaneous closure of their VSD. Unoperated patients with an isolated small VSD and normal PVR have an excellent long-term prognosis, although they remain at risk for endocarditis (65,76). Unoperated patients with moderate to large shunts are at risk for multiple complications, including endocarditis, aortic regurgitation, LV dysfunction from chronic volume overload, arrhythmias, development of the Eisenmenger syndrome, and sudden death. Patients with subarterial VSDs (and occasionally perimembranous defects) may develop prolapse of the aortic cusp through the defect with the development of progressive aortic regurgitation (65,71,77).
Overall, late outcome after early surgical closure of a VSD is excellent. Residual shunts are common, seen in up to 20% of cases after surgery, but are usually small (78). Late complications after surgical repair include endocarditis (if a residual shunt persists after surgery), surgically induced aortic or pulmonary regurgitation, and tricuspid regurgitation (if the septal leaflet was manipulated during the VSD repair). Arrhythmias and conduction disturbances may be seen (79,80). Right-bundle-branch block occurs in 30% to 60% of patients after surgical closure, first-degree AV block is seen in 10%, and complete heart block in 1% to 3% over long-term follow-up (71,78). Patients may have LV dysfunction with late repair of the defect or with significant aortic regurgitation. Patients may have persistent pulmonary hypertension after surgery or may develop progressive pulmonary hypertension despite successful closure of their shunt (65). There is an increased risk of sudden cardiac death after VSD closure, seen in 2% of patients (65,71). The etiology for sudden death has not been defined.
In general, patients undergoing early repair without a residual shunt, evidence of pulmonary hypertension, arrhythmias, or conduction block do not require long-term follow-up. Later repair of VSDs is associated with a risk of pulmonary hypertension and LV dysfunction, making long-term follow-up of these patients mandatory.
Patent Ductus Arteriosus
Patent ductus arteriosus (PDA) refers to continued patency of a normal structure in fetal circulation (Fig. 30.3). The ductus arteriosus is derived from the left sixth aortic arch and is usually a left-sided structure, but may be right sided or bilateral. PDA occurs in 5% to 10% of congenital defects and is often present in premature infants (3,81,82). An increased incidence is also seen in cases of maternal rubella.
 FIGURE 30.3. Patent ductus arteriosus with left-to-right shunting. (From Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. N Engl J Med 2000;342:340. ) |
A PDA is associated with a left-to-right shunt that is predominantly systolic in early infancy but becomes continuous as PVR decreases. Thus, in the newborn with a significant shunt, there is an active precordium and only a systolic murmur. In older children and adults, a continuous “machinery” murmur is present at the left upper sternal border. A PDA may close spontaneously before the age of 6 months.
The clinic presentation of a PDA is similar to that of a VSD. With small shunts, there is a loud murmur but the ECG and CXR are normal. In patients with a moderate to large shunt, the clinical exam is remarkable for an enlarged apical impulse and bounding pulses in addition to the continuous murmur from the shunt. With a moderate to large shunt, the ECG may show LVH and the CXR will demonstrate cardiac enlargement and pulmonary plethora. Patients with moderate to large shunts may develop atrial arrhythmias, left heart failure, and pulmonary hypertension, including the development of the Eisenmenger syndrome. In patients with the Eisenmenger syndrome, the continuous murmur disappears as the aortic-to-pulmonary pressure gradient decreases. Patients with Eisenmenger syndrome secondary to a PDA have preferential shunting of deoxygenated blood to the descending thoracic aorta through the ductus while oxygenated blood is ejected into the ascending aorta. This leads to the differential cyanosis, with cyanosis and clubbing in the feet but not in the upper extremities. Because of the proximity of the left subclavian artery to the ductus, there may be some clubbing and cyanosis in the left hand. All patients are at risk for endarteritis, regardless of the size of the shunt. An exception to this rule is the “silent ductus,” which refers to a trivial shunt seen by color flow Doppler without an associated murmur. A “silent ductus” has no hemodynamic significance, and the risk of endocarditis is extremely low (83).
Echocardiography demonstrates continuous flow from the aorta to the pulmonary artery by color Doppler. Direct visualization of the ductus by echocardiography is usually possible in children but difficult in adults. The aortic-to-pulmonary artery pressure gradient can be measured from the continuous-wave Doppler tracings. The presence of other associated congenital defects should be a routine part of the exam. The diagnosis can also be made and/or confirmed at cardiac catheterization by the presence of an oxygen saturation step-up at the level of the pulmonary artery and demonstration of the ductus by aortography.
In premature infants, closure of the ductus arteriosus can be promoted with the use of indomethacin (once coarctation and ductal-dependent congenital defects are excluded) (84). Closure of the ductus is generally recommended for all shunt sizes (except silent PDA) to reduce the risk of endarteritis as well as that of LV volume overload in moderate to large shunts (85). Closure of a PDA is contraindicated in the setting of Eisenmenger physiology.
Closure of a PDA can usually be accomplished with percutaneous closure devices. The Rashkind double umbrella device was introduced in 1979 and had an occlusion rate of 82.5% at 1 year and 94.8% at 20 months but a fairly high rate of complications. The device is not currently approved by the Food and Drug Administration (FDA) (86). Coils may be used to close a PDA. Coils have been available since 1976 (never applied for FDA approval) and can be deployed using small delivery systems. Closure rates of 93% to 97% have been reported with coil embolization after 6-month follow-up (87,88). Embolization into the pulmonary circulation was reported in 3% to 8% of initial studies (87,88,89), but the incidence is lower after modifications in technique (90,91,92,93). The Amplatzer duct occluder has shown excellent early outcomes, with a 98% closure rate at 6 months (94). The U.S. experience with the Amplatzer device reported in 2004 demonstrated a 99% success rate for device implant, with 76% occlusion on initial angiogram, 89% occlusion by postprocedure day 1, and 99.7% occlusion at 1 year. Serious adverse events were seen in 2.3% of cases (95). Patients after device closure still require bacterial endocarditis prophylaxis for at least 6 months, lifelong if there is a residual shunt. There are rare reported cases of hemolysis after device implant (96,97).
Surgical ligation and division of the ductus can be done with low morbidity and mortality but is rarely needed (98,99,100). Surgical closure is required for a PDA that is too large for device closure and for those patients with distorted ductal anatomy (e.g. aneurysm or calcification).
A patient with a PDA repaired in childhood can be considered “cured.” Patients repaired in adolescence or adulthood remain at risk for complications such as pulmonary hypertension, LV dysfunction, and arrhythmias and should have routine follow-up. Although rare, an aneurysm of the PDA may occur with risk of rupture. The natural history of device closure is unknown. Intermittent follow-up is advisable.
Atrioventricular Septal Defects
Partial and complete atrioventricular septal defects (AVSDs) are seen in 2% to 3% of patients with congenital heart disease. Also known as AV canal defects or endocardial cushion defects, they are characterized by a common atrioventricular junction guarded by a common atrioventricular valve. There is absence of the atrioventricular septum that separates the RA from the LV. The aorta is “unwedged” from its usual position between the atrioventricular orifices, which results in a narrowing of the subaortic region and a longer outflow dimension of the interventricular septum. There is considerable variation in the features of AVSDs, including variations in the common
atrioventricular valve, differences in the degree and direction of shunting, and the relative “balance” of the atrioventricular valves and the ventricles (101). With a partial atrioventricular septal defect (partial AVSD), the common AV valve is divided into separate right and left orifices, which are separated by fusion between the superior and inferior bridging leaflets. This gives a three-leaflet appearance to the mitral valve, often mistaken as a cleft in the anterior leaflet. On echocardiography, the mitral and tricuspid valves appear at the same level at the AV junction. Partial AVSDs may have attachment of the bridging leaflets of the common AV valve to the ventricular septum, resulting in only interatrial shunting (the so-called ostium primum defect). Alternatively, the bridging leaflets may be attached to the atrial septum, resulting in only an interventricular shunt (inlet-type VSD).
atrioventricular valve, differences in the degree and direction of shunting, and the relative “balance” of the atrioventricular valves and the ventricles (101). With a partial atrioventricular septal defect (partial AVSD), the common AV valve is divided into separate right and left orifices, which are separated by fusion between the superior and inferior bridging leaflets. This gives a three-leaflet appearance to the mitral valve, often mistaken as a cleft in the anterior leaflet. On echocardiography, the mitral and tricuspid valves appear at the same level at the AV junction. Partial AVSDs may have attachment of the bridging leaflets of the common AV valve to the ventricular septum, resulting in only interatrial shunting (the so-called ostium primum defect). Alternatively, the bridging leaflets may be attached to the atrial septum, resulting in only an interventricular shunt (inlet-type VSD).
With a complete atrioventricular septal defect (complete AVSD), the common AV valve “floats” between the atrial and ventricular septum, allowing shunting at both atrial and ventricular levels. Patients with a complete AVSD may have a common valve with a single orifice or may have separate left and right orifices. When the common AV valve is equally committed to both ventricles, this is referred to as a balanced defect. In unbalanced forms, there is commitment of the common AV valve to one ventricle with resultant hypoplasia of the other ventricle. Many other associated malformations may coexist, including left ventricular outflow obstruction, other congenital deformities of the AV valve, and association with other forms of congenital heart disease (e.g., tetralogy of Fallot, double-outlet right ventricle, etc.). There is a distorted arrangement of atrioventricular node (located in the posterior atrial wall) and the bundle of His (located under the inferior bridging leaflet), which makes these patients prone to conduction block.
AVSDs are particularly common in patients with Down syndrome (seen in 30% to 40% of Down patients with CHD), usually as the complete form of AVSD. Conversely, Down syndrome is present in 70% to 80% of patients with a complete AVSD (10,11). The risk of developing associated pulmonary vascular disease appears to be greater and more rapidly progressive in patients with Down syndrome (102). This is due, at least in part, to a tendency toward airway obstruction (due to macroglossia, a small hypopharynx, and poor pharyngeal muscle tone), abnormal capillary bed morphology in the lungs, and possible pulmonary hypoplasia (103,104).
The clinical presentation depends on the specific malformation (105). Partial AVSDs of the ostium primum type present similar to other ASDs, except for the unusual QRS axis (leftward and superior counterclockwise axis). Patients may have first-degree AV block as well. Partial AVSDs of the inlet VSD type present similar to other VSDs. Patients with complete AVSDs typically present with breathlessness and heart failure in infancy due to excessive pulmonary blood flow. Symptoms usually becomes manifest after PVR falls, usually within a few weeks of birth. These patients are also at significant risk for pulmonary hypertension if not repaired. The magnitude of left-to-right shunting and the degree of regurgitation through the common AV valve determine the clinical presentation. Patients with large shunts and/or significant AV valve regurgitation present earlier with symptoms of heart failure and/or evidence of pulmonary hypertension. Varying degrees of cyanosis may be present if there is significant right-to-left shunting.
The ECG in patients with a complete AVSD shows left-axis deviation due to an abnormal activation sequence of the ventricles (due to deficiency of intermediation radiation of left bundle branch). First-degree AV block is common, and right-bundle-branch block and right ventricular hypertrophy (RVH) are invariably present. Left ventricular hypertrophy (LVH) may be present as well. The CXR shows cardiomegaly and pulmonary plethora.
Echocardiography places a critical role in the diagnosis, including the anatomy and function of the common AV valve, the location and degree of intracardiac shunts, the balance of the ventricles, and the presence of other associated conditions (106). Cardiac catheterization with angiography is often warranted to assess hemodynamic parameters and the magnitude of the intracardiac shunts. In patients with significant pulmonary hypertension, a lung biopsy is occasionally required to determine operability.
Surgical repair should be undertaken at the time of diagnosis if pulmonary vascular disease is not prohibitive. Surgical repair of complete AV septal defects is complex, requiring closure of the shunts and creation of two competent AV valves (107,108). In some cases, a pulmonary artery band is placed to prevent pulmonary hypertension if repair of the defect cannot be performed.
When diagnosis of made in adolescence or adulthood, there is usually a partial AV septal defect or the patient has developed severe pulmonary vascular obstructive disease. Patients who present in adulthood with a partial AVSD are usually candidates for surgery (109). Device closure is not an option. Patients with a complete AVSD and severe pulmonary hypertension should be treated as having Eisenmenger physiology once the diagnosis is confirmed.
The long-term outcome after surgical repair is good (110,111). All patients are at risk for endocarditis and require antibiotic prophylaxis. Despite a good prognosis after surgery, patients remain at risk for left-sided AV valve regurgitation and need long-term follow-up. Surgical series have indicated that as many as 10% to 12% of patients will need further surgery for left-sided AV valve regurgitation. Patients may also have a residual VSD, may develop subaortic or subpulmonary obstruction, or may develop progressive pulmonary vascular disease (especially with “late” closure of VSD). Patients are also at risk for development of complete heart block, sinus node dysfunction, and atrial arrhythmias. Sudden death has been reported, although the risk is largely unknown.
Aortopulmonary Window
The aortopulmonary window is a rare form of congenital heart disease, manifested by a communication between the ascending aorta and the pulmonary trunk above the level of the coronary arteries (112,113). An aortopulmonary window can occur as an isolated defect but is often associated with other anomalies. The defect is usually very large, resulting in a large left-to-right shunt and a high likelihood of developing pulmonary hypertension. Thus, patients usually present in infancy with symptoms of heart failure or with cyanosis due to pulmonary hypertension with right-to-left shunting. The diagnosis is made by echocardiography demonstrating the defect and the associated shunting. Occasionally, defects may be small enough to be closed with catheter intervention (114), but surgical closure is required for the majority (115,116,117). Presentation in adulthood is nearly always associated with the Eisenmenger syndrome.
Partial Anomalous Pulmonary Venous Connection
Partial anomalous pulmonary venous connection is defined as one or more (but not all) pulmonary veins connecting to a systemic vein, the RA, or the coronary sinus. Examples include connection of the right upper lobe and right middle lobe veins to the SVC, right upper lobe and right middle lobe veins to the RA, right pulmonary veins to the IVC, right lower lobe vein to the IVC (scimitar syndrome), left upper or lower pulmonary veins to the coronary sinus, and left lower pulmonary veins to the RA or IVC (118,119,120).
Partial anomalous pulmonary venous connection is relatively uncommon, accounting for less than 1% of all congenital defects. An ASD is usually present, and the clinical
presentation is similar to that of an uncomplicated ASD. Partial anomalous pulmonary venous connection can be seen with any type of ASD but is most commonly associated with the sinus venosus type of ASD. Other associated defects may occur. The exam findings, ECG, and CXR findings in patients with partial anomalous pulmonary venous connection are similar to those of a secundum ASD. The CXR findings in the scimitar syndrome are addressed later.
presentation is similar to that of an uncomplicated ASD. Partial anomalous pulmonary venous connection can be seen with any type of ASD but is most commonly associated with the sinus venosus type of ASD. Other associated defects may occur. The exam findings, ECG, and CXR findings in patients with partial anomalous pulmonary venous connection are similar to those of a secundum ASD. The CXR findings in the scimitar syndrome are addressed later.
Patients are usually asymptomatic in childhood. They may remain undiagnosed in adulthood if the anomalous pulmonary venous connection is an isolated defect. If more than 50% of the total pulmonary blood flow drains to the right heart, symptoms are common. Thus, symptomatic patients usually have more than one anomalous connection or an associated lesion. Symptoms are similar to those of ASD, with dyspnea, arrhythmias, and (rarely) pulmonary hypertension. The diagnosis can be made by echocardiography if care is taken to identify the pulmonary vein connections. TEE is usually required in adult patients to adequately define the pulmonary vein anatomy (121). Cardiac MRI is an excellent tool for the diagnosis of partial anomalous pulmonary venous connections (122).
Treatment depends on the magnitude of shunting. Isolated anomalous pulmonary venous connection with a small shunt does not require surgery. For larger shunts, surgical closure of the ASD and rerouting of pulmonary venous return to the left atrium is performed to prevent long-term complications such as atrial arrhythmias, right heart failure, and pulmonary hypertension (123,124). Pulmonary venous drainage should be assessed in any patient with an ASD who is being considered for either device or surgical closure. The presence of an anomalous pulmonary venous connection is a contraindication to device closure. Inspection of the pulmonary venous connections should be a routine part of surgical closure of an ASD, and anomalous pulmonary venous connections should be repaired when present.
The long-term outcome after surgical repair of partial anomalous pulmonary venous connection is excellent. Bacterial endocarditis prophylaxis is not required after surgical repair. Obstruction of the reimplanted pulmonary veins or obstruction of the vena cava at the site of pulmonary vein explantation is uncommon but may require surgical or catheter intervention (127).
Scimitar Syndrome
The scimitar syndrome refers to the presence of anomalous drainage of the right pulmonary veins to the IVC with a characteristic appearance on CXR resembling a scimitar or Turkish sword. There is usually some degree of hypoplasia of the right lung and the right pulmonary artery, usually with an aberrant systemic artery from thoracic aorta supplying part of the right lung. Surgical correction removes the left-to-right shunt and may improve blood flow to the right lung. There is a risk of postoperative pulmonary venous obstruction (126).
Obstructive Lesions
Pulmonary Stenosis
Pulmonary stenosis (PS) is a common defect, occurring in 7% to 10% of patients with congenital heart disease. Obstruction may be subvalvular, valvar, or supravalvular. Valvar stenosis is the most common (90%). The pulmonary valve morphology varies. The valve may be unicommissural with an eccentric orifice (extremely rare), bicuspid or trileaflet with commissural fusion, or dysplastic. Dysplastic valves have markedly thickened leaflets with disorganized myxomatous tissue but minimal commissural fusion. Bicuspid or trileaflet valves with commissural fusion are usually amenable to balloon dilation or surgical valvotomy, whereas dysplastic valves are less amenable to these procedures. Dysplastic valves are commonly associated with Noonan syndrome (127). PS is usually an isolated lesion. Associated cardiac and noncardiac malformations are more common when the valve is dysplastic (128). Chronic obstruction of the right ventricular outflow tract leads to RVH, which may be particularly prominent in the infundibular region, further contributing to RV outflow tract obstruction. Severe PS presenting in infancy is associated with severe RVH and a small right ventricular cavity size. Lesser degrees of PS are associated with RVH, but the RV cavity is usually well formed. The degree of stenosis is classified by peak systolic gradient, with trivial stenosis defined as a peak gradient less than 25 mm Hg, mild stenosis with a gradient of 25 to 49 mm Hg, moderate stenosis with a gradient of 50 to 79 mm Hg, and severe stenosis with a peak gradient above 80 mm Hg.
Infants with critical PS present in neonatal period, usually with cyanosis due to right-to-left shunting across a PFO or ASD. Mortality is high in neonates with critical PS unless intervention is prompt (129). Lesser degrees of stenosis usually present later in childhood or in adulthood. Patients with trivial or mild stenosis with a peak gradient of less than 25 mm Hg have a good outcome. There is usually no significant progression of disease, and therefore no treatment is warranted. Patients with more significant stenosis may present with exertional dyspnea, chest pain, fatigue, or syncope, occasionally with cyanosis (130).
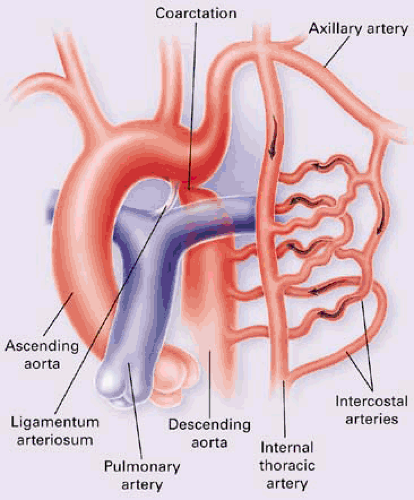 FIGURE 30.4. Coarctation of the aorta with development of extensive collaterals (internal mammary and intercostal arteries). (From Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. N Engl J Med 2000;342:340. ) |
The physical exam findings in valvar PS include an RV lift, a thrill along the left sternal border, and a harsh crescendo-decrescendo systolic ejection murmur in the pulmonary area, which is louder in expiration. A systolic ejection click is often present. The intensity of P2 is reduced in patients with severe stenosis. The ECG reflects the degree of RVH (except in the neonatal period). The ECG is normal with mild degrees of obstruction and shows right-axis deviation and RVH with moderate to severe obstruction. Poststenotic dilation of the main pulmonary artery and the left pulmonary artery (due to a more parallel take-off of the left pulmonary artery) may be seen on CXR with all degrees of PS (131,132). Heart size is normal with mild to moderate obstruction, but right-sided enlargement is seen with severe stenosis. Echocardiography is the diagnostic method of choice and can demonstrate valvular and infundibular anatomy. Continuous-wave Doppler is used to quantitate the transvalvular gradient. A complete study includes an assessment of the integrity of atrial septum as well as assessment of RV size and function.
In the Second Natural History Study of Congenital Heart Defects, patients with mild PS (peak gradient 25 to 49 mm Hg) had a 20% chance of requiring intervention at some point. Patients with moderate stenosis treated medically were at risk for progressive obstruction with symptoms warranting intervention. Most patients with a peak gradient greater than 50 mm Hg required intervention, with better outcomes demonstrated in those patients undergoing intervention than in those treated medically (132).
The earliest surgical interventions on the pulmonary valve were performed using a closed technique with blunt dilation of the right ventricular outflow tract (Brock procedure). Currently, pulmonary valvotomy is performed using cardiopulmonary bypass (133). Transcatheter intervention with balloon valvuloplasty has now largely replaced operative intervention and has become the therapy of choice (134,135,136,137). Balloon valvuloplasty can be performed with a low rate of complications and outcomes similar to surgery with a similar reduction in gradient (138). An infundibular gradient may be present after successful pulmonary valvotomy but often regresses over 3 to 12 months.
Long-term outcomes for both surgical and balloon valvotomy are excellent. Long-term complications of both procedures include pulmonary regurgitation and residual or recurrent RV outflow tract obstruction. Reintervention is required in some cases for recurrent RV outflow obstruction with symptoms or significant arrhythmias. The presence of severe pulmonary regurgitation with decreasing exercise capacity, deteriorating RV function, or the development of significant arrhythmias is an indication for pulmonary valve replacement (133).
Subpulmonary stenosis is usually seen in complex defects, such as tetralogy of Fallot. Isolated subpulmonary stenosis is rare.
Supravalvular obstruction with pulmonary arterial stenosis may be seen in rubella syndrome and Williams syndrome and in association with other complex congenital cardiac defects (e.g., tetralogy of Fallot). Pulmonary arterial stenosis may also occur as an isolated defect (139,140). Stenosis may occur in the main pulmonary artery, at the bifurcation of the pulmonary artery branches, and at the secondary or more distal branches. The obstruction may be focal or diffuse (often a manifestation of more widespread vasculopathy such as rubella, cutis laxa, or Ehlers-Danlos syndrome) (141).
The clinical presentation is similar to that of valvar PS. Patients have a systolic ejection murmur, but there is no ejection click, and P2 is normal. The ECG and CXR findings are usually nonspecific. Echocardiography may demonstrate the presence of RV pressure overload and may demonstrate the site of stenosis, but the branch pulmonary arteries may be difficult to visualize by TTE. Doppler study of the main and branch pulmonary arteries is used to quantitate the severity of stenosis. CT or MRI scans offer excellent visualization of the main pulmonary artery trunk and the pulmonary artery branches (142). Angiography can also be used to demonstrate the obstruction, and the localized pressure gradient can be demonstrated at catheterization. Perfusion imaging helps in assessing perfusion imbalance. Although pulmonary artery stenosis may be managed surgically with pericardial or prosthetic patch repair to enhance stenotic vessels, stenting of pulmonary vessels is playing an increasing role (143,144,145,146).
Obstruction of the Left Ventricular Outflow Tract
Left ventricular outflow tract obstruction may occur at the level of the valve, below the valve, or in the ascending aorta. Valvar aortic stenosis (AS) is the most common cause of congenital LV outflow obstruction, and bicuspid valves are by far the most common type. The bicuspid aortic valve is usually not included in epidemiologic studies of congenital heart disease but is the most common type of congenital cardiac defect, seen in 1% to 2% of the general population (147,148,149). A bicuspid aortic valve results from fusion of two commissures, resulting in two rather than three valve leaflets. There is a high incidence of bicuspid valves in a mouse model of nitric oxide synthase deficiency, suggesting a role of nitric oxide in the development of the normal trileaflet aortic valve (150). Bicuspid aortic valves may be familial, but most appear to be spontaneous mutations. Bicuspid valves and coarctation of the aorta are the most common defects found in patients with Turner syndrome (XO) (151).
Considered a normal variant by some, a bicuspid aortic valve may function normally throughout life or may develop either stenosis or regurgitation. There are autopsy reports of normally functioning bicuspid valves in octogenarians (147,148). The abnormal opening of the valve leaflets is presumed to cause an abnormal flow profile across the valve, resulting in valve thickening, fibrosis, and calcification. This, in turn, may result in either progressive stenosis or regurgitation. Although it is usually an isolated abnormality, associated cardiac defects are seen in up to 20% of patients (including coarctation of the aorta, PDA, and VSD). Left-dominant coronary circulation is seen in 30% to 60% of cases. Patients with bicuspid aortic valves may have associated abnormalities within the media of the aorta, placing them at risk for aortic root dilation and dissection (149,152). The degree of aortic root abnormality may be out of proportion to the severity of the valvular dysfunction (153).
Congenital AS presenting in infancy is unusual (occurs in 10% to 15% of patients with congenital AS) and may be associated with other lesions. The valve morphology in isolated valvar AS in the neonate may be bicuspid, unicuspid, or severely dysplastic. Neonates with severe AS typically present with severe decompensation, and prompt intervention is required (154,155).
Children with less severe degrees of AS are usually asymptomatic, and the diagnosis is usually made based on the presence of a murmur. Bicuspid aortic valves are often seen in adults as an incidental finding.
Children and adults with congenital AS usually have a systolic ejection click and a systolic ejection murmur. The aortic closure sound (A2) may be normal or decreased due to decreased leaflet mobility. The second heart sound may be normal or narrowly split. Paradoxical splitting of the second heart sound may be present with severe obstruction. The ECG may be normal or may demonstrate LVH. There is poor correlation between the degree of stenosis and the ECG findings. The CXR may demonstrate a normal heart size or cardiomegaly. Poststenotic dilation of the ascending aorta may be seen.
The clinical manifestations of AS are similar for congenital and acquired forms. Chest pain, congestive heart failure symptoms, and syncope (often exertional or postexertional) are the presenting symptoms. Unlike acquired forms of AS, symptomatic congenital AS tends to present earlier in life (in the forties or fifties). In the adult population, bicuspid aortic valves account for half of all surgical cases of isolated AS. Patients with associated abnormalities of the aortic media may present with aortic root dilation, dissection, and rupture. All patients are at risk for endocarditis, particularly those with bicuspid aortic valves.
Echocardiography is the diagnostic tool of choice for the diagnosis of AS (156). Patients with a bicuspid aortic valve typically have two unequal-size aortic cusps with an eccentric closure line. Valve anatomy can be determined, and the presence of associated regurgitation or stenosis can also be demonstrated and quantitated, with assessment of the transvalvular gradient, and calculation of valve area. LVH and LV function (both systolic and diastolic) should be assessed.
Cardiac catheterization is usually reserved for patients in whom either surgical or catheter intervention is contemplated or for additional quantitation of the severity of valvular disease. Coronary angiography is usually performed in patients older than the age of 40 years. Visualization of the aortic root by echo, angiography, or MRI is an important part of the diagnostic evaluation. All patients with congenital AS need bacterial endocarditis prophylaxis. The indications for catheter or surgical intervention differ between the pediatric and adult populations. In children, there is a risk of sudden death in the absence of definable symptoms related to the aortic valve disease. For this reason, catheter or surgical intervention is recommended for those patients with severe stenosis as defined by a valve area of less than 0.5 cm2/m2 or a mean gradient of greater than 50 mm Hg (157). Exercise testing is also used, with the development of ST-segment depression or significant ventricular ectopy considered to be a reasonable indication for intervention. For mild stenosis (gradient <40 mm Hg), observation is recommended because the risk of sudden death is low.
In the adult population, the indications for surgical intervention are the same as for those patients with acquired forms of AS, namely chest pain, congestive heart failure, and syncope.
Unlike the pediatric population, sudden death occurs rarely in asymptomatic adult patients with AS (158). In symptomatic adults, survival is poor without surgical intervention, with 5-year survival rates ranging from 15% to 50%. Because of the associated aortic root abnormalities, the initial presentation of a patient with a bicuspid aortic valve may be in the setting of aortic dissection or rupture (149,153).
Unlike the pediatric population, sudden death occurs rarely in asymptomatic adult patients with AS (158). In symptomatic adults, survival is poor without surgical intervention, with 5-year survival rates ranging from 15% to 50%. Because of the associated aortic root abnormalities, the initial presentation of a patient with a bicuspid aortic valve may be in the setting of aortic dissection or rupture (149,153).
In the pediatric and adolescent populations, balloon valvotomy is a reasonable option for many patients. Successful balloon valvotomy requires a pliable, noncalcified valve. Surgical options in the pediatric age group include valvotomy and valve replacement. The Ross procedure is usually preferred in the pediatric population because of the potential for growth of the “neoaortic” valve in proportion to patient’s somatic growth. In adolescents and young adults, balloon valvotomy may be an option if the valve appears pliable and has little or no calcification. In most adult patients, there is usually significant calcification and leaflet thickening, making balloon and surgical valvotomy unattractive. Thus, aortic valve replacement is usually required. Late complications of both balloon valvotomy and surgical valvotomy are well recognized. Series of surgical valvotomy have similar outcomes to catheter-based procedures, with a 40% to 50% incidence of reoperation for recurrent stenosis or regurgitation (159,160,161,162,163,164,165,166,167,168). Aortic regurgitation is common postvalvuloplasty (either surgical or balloon). The degree of aortic regurgitation (AR) is usually mild, but it is moderate in 20% to 30% of cases, with a potential to progress over time. LV dysfunction may occur, usually in patients with a later age at repair and/or a history of prolonged, severe obstruction. The reported risk of sudden death in the postsurgical valvotomy population is 0.4% per year, usually associated with residual or progressive aortic valve disease (169). All patients remain at risk for endocarditis.
Subaortic stenosis may take many forms, ranging from a discrete fibrous or muscular ridge to a more diffuse (tunnel) form of muscular hypertrophy (170,171,172,173,174). Subaortic obstruction may be isolated or may occur in association with other defects (in 60%). The most common associated defects are VSD, coarctation, Shone syndrome, PDA, and valvar AS. Subaortic obstruction occurs due to an accumulation of fibroelastic tissue and may be an acquired lesion. Subtle abnormalities of the LV outflow tract may result in altered shear stress, triggering cell proliferation (175). Progression of stenosis is common, although the rate of progression is variable and often difficult to predict (176,177,178,179,180). Downstream turbulent flow may result in damage to the aortic leaflets, resulting in aortic regurgitation in as many as 50% of patients (181).
The clinical presentation varies. Patients with mild obstruction are usually asymptomatic. Dyspnea, chest pain, or syncope is seen with moderate to severe obstruction. The diagnosis is made by echocardiography demonstrating narrowing of the LV outflow tract. A discrete membrane may be visualized, or there may be more diffuse narrowing and obstruction. Measurement of the severity of subaortic stenosis by Doppler is accurate, with discrete forms of obstruction, but it may be inaccurate in long, tunnel-like stenosis. Cardiac catheterization is used to measure the subaortic gradient and to visualize the subvalvular anatomy by angiography. Management is controversial for asymptomatic patients. Patients are at risk for progressive obstruction and progressive aortic valve damage. Some experts suggest waiting for symptoms, whereas others propose early intervention to prevent aortic valve damage (182,183,184,185). In general, a resting gradient greater than 50 mm Hg and/or progressive AR are indications for surgery. Surgery is more complicated for tunnel-type stenosis, which has a higher operative mortality (186). In addition to resection of subaortic tissue, some patients may need augmentation of the LV outflow tract (aortoventriculoplasty or Konno procedure). These patients are at risk for complete AV block as a complication of surgery. All patients are at risk for progressive AR, even after successful relief of subaortic obstruction, which occurs in as many as 25% to 40% of cases. Recurrence of LV outflow tract obstruction after successful surgical excision occurs frequently, and is reported to be as high as 27% (182,183,184,185,186,187).
Supravalvular AS is the rarest form of LV outflow tract obstruction, and is caused by a variety of different pathologic lesions. All forms of supravalvular obstruction tend to progress over time. Supravalvular AS is frequently seen in Williams syndrome (supravalvar AS, intellectual impairment, and distinct facial features) (188,189). Supravalvular stenosis may also occur as an isolated sporadic case and occasionally in familial form. The most common manifestation is narrowing of aorta at the level of the sinotubular junction. There is potential for involvement of the coronary artery ostia, including dilation of coronary arteries and obstruction (190,191), aneurysms of the ascending aorta (192), pulmonary artery stenosis (193), and involvement of other major arterial branches, including the cerebral circulation (194,195). The aortic valve is abnormal in 35% to 50% of cases, with bicuspid valves and aortic regurgitation or stenosis. The diagnosis of supravalvar obstruction may be made by echocardiography, MRI, or catheterization.
Surgery is performed for all symptomatic patients. Other indications for intervention include a gradient of greater than 50 mm Hg or progressive aortic valve dysfunction in asymptomatic patients. Surgery involves relieving obstruction while preserving aortic root geometry and aortic valve function. The Ross procedure is often recommended for those patients requiring aortic valve replacement. Recurrence of supravalvular stenosis is uncommon after repair. Reoperation is required in 17% to 40% of patients undergoing surgery at an early age, usually aortic valve replacement for progressive AR (196,197,198,199).
Coarctation of the Aorta
Coarctation of the aorta is defined as a narrowing or obstruction of the aortic arch. Coarctation is a common defect and occurs in 8% to 10% of all congenital defects; it is more common in male than in female individuals (2:1) (33,200). Aortic coarctation may occur in isolation but is often associated with other congenital defects (bicuspid aortic valve in up to 85% of cases; also VSD and mitral valve abnormalities). Aortic coarctation is common in Turner syndrome, occurring in 30% of cases.
Aortic obstruction usually occurs at the level of the ligamentum arteriosus and is due to thickened intima and increased tissue in the media consisting of collagen, smooth muscle cells, and varying degrees of elastin. A more diffuse arteriopathic process appears to be involved because some patients have a propensity to aortic aneurysm formation and dissection or may have associated aneurysms in the circle of Willis (seen in 10% of cases) (201).
The clinical presentation depends on the location and severity of the obstruction. More than half of patients present with symptoms in the first year of life. In the infantile type, systemic blood flow depends on flow through the ductus to the descending thoracic aorta. In these infants, ductal closure can result in circulatory collapse. The use of prostaglandin E1 to maintain ductal flow is a temporizing measure, and immediate intervention is required due to the absence of adequate collateral circulation. After repair in infancy, these patients remain at risk for premature atherosclerosis, late hypertension, and premature death (202,203,204).
Presentation in older children and adults is different. In these patients, blood flow to the descending thoracic aorta is supplied by the LV through the ascending aorta. Collateral circulation gradually develops between the proximal and distal aorta (Fig. 30.4). These patients are usually asymptomatic and present with upper limb hypertension.
The physical exam findings depend on the age of presentation. In the infantile form, the infant is usually in circulatory shock and may have differential cyanosis. In adolescents and adults, the exam is remarkable for a blood pressure difference between the upper and lower extremities and diminished or absent femoral pulses. A short systolic murmur from the coarctation is common and may be heard in the left interscapular area. Faint, continuous murmurs from collateral vessels may also be audible. The ECG is often normal but may show LVH. On CXR, the heart size may be normal or mildly enlarged. Rib notching from the fourth to eighth ribs may be seen in older children and adults due to hypertrophied intercostal arteries as part of the collateral circulation (205). A “3” sign representing a pre- and poststenotic dilation of the aorta at the level of the coarctation may be seen.
Echocardiography is a good technique for the diagnosis of coarctation, demonstrating the area of obstruction in the aorta and demonstrating disturbed flow by Doppler techniques. Continuous-wave Doppler with the expanded Bernoulli equation is needed to accurately assess the degree of obstruction. For patients with severe stenosis and extensive collaterals, the Doppler gradient may underestimate the degree of obstruction due to decreased blood flow through the coarctation segment (206). MRI is excellent for demonstrating aortic anatomy and is particularly useful in the adult population in whom echocardiographic imaging of the aortic arch and descending thoracic aorta may be difficult (207,208).
The long-term outcome for patients with coarctation of the aorta is poor without intervention. Sixty percent of patients with symptomatic coarctation and 90% with complicated coarctation (associated with other lesions) will die within the first year of life without intervention (209). The average life expectancy for simple coarctation without surgery is 35 years. Presentation in adulthood suggests mild to moderate postductal coarctation.
Indications for intervention beyond the neonatal period include congestive heart failure, the presence of upper extremity hypertension, and/or a gradient greater than 20 mm Hg across the obstruction. Exercise testing may be used to provoke a gradient across the area of obstruction.
Surgery for “native” coarctation in children can be done with an end-to-end anastomosis, a subclavian flap (to augment the aortic arch), or an interposition graft (210,211,212). In adults, resection of the obstructed segment with end-to-end anastomosis is the procedure of choice (213,214,215). An interposition tube graft may be needed if there is a long segment of coarctation. A bypass jump graft is occasionally required in older patients with fragile aortic tissue or a long segment of obstruction. Postoperative complications include hypertension, abdominal pain, chylothorax, late aneurysm formation, and, rarely, spinal cord ischemia.
Percutaneous transcatheter angioplasty of native or recurrent coarctation has an increasing role. In native coarctation, balloon procedure with or without stent placement is a treatment alternative. Acute and long-term results for native aortic coarctation are similar to those of surgery (216,217,218,219,220,221), but aortoplasty is associated with higher rates of aneurysm formation and restenosis than surgery. Balloon aortoplasty appears to be a better option than surgery for recurrent coarctation (222,223,224). Complications of catheter procedures include femoral artery injury and thrombosis, aneurysm formation, embolic events, and, rarely, aortic rupture. Hypertension is common, even after successful relief of obstruction (202,225,226,227). Hypertension is seen in as many as 75% of patients after repair and is more common in those patients with older age at repair. The incidence of hypertension appears to increase with longer follow-up. Patients with normal resting blood pressure often demonstrate an abnormal blood pressure response to exercise as well as increased left ventricular mass. Although the pathophysiology is not well understood, the hypertension is likely to be related to structural changes in the central and peripheral vessel walls, abnormalities of endothelial reactivity, and alterations in the renin–angiotensin system. In some patients, there is persistent hypoplasia of the aortic arch, which contributes to persistent hypertension. Chronic hypertension places patients at risk for premature coronary artery disease (CAD), LV dysfunction, rupture of aortic or cerebral aneurysms, and sudden death. Meticulous blood pressure control is mandatory. β-Blockers are usually recommended as first-line therapy, although there are no randomized trials.
Lifelong follow-up with imaging of the aorta is mandatory but not often employed. Even after successful “repair,” there is evidence of ongoing morbidity and mortality in these patients (203,228). In one long-term follow-up of postoperative coarctation repair, the overall 30-year survival was only 72%. Thirteen percent of patients required reoperation for either aortic valve replacement or recurrent coarctation (228). Residual or recoarctation may be seen in 3% to 41% of patients and can occur with any surgical technique or after angioplasty (seen in 8% to 11% of patients undergoing angioplasty for native coarctation). Residual or recurrent obstruction is associated with hypertension, increased LV mass, and the development of CAD and congestive heart failure (229). Angioplasty is usually recommended for recoarctation after previous surgery, with a good success rate (65% to 100%) and an acceptable (13%) complication rate (222,223,224). Stents are increasingly being used for recurrent coarctation, although only short-term data are available (230,232).
Patients may have aneurysm formation at site of repair (seen in 5% to 9% of surgical patients and 4% to 12% of patients after balloon procedures) and are also at risk for dissection and rupture. Both repaired and unrepaired patients may have evidence of a diffuse arteriopathy of the aorta of unclear etiology and may develop aneurysm formation and dissection at a site
remote from the original site of coarctation (201). All patients (repaired and unrepaired) are at risk for endarteritis or endocarditis on an associated bicuspid valve. As many as 10% of patients with coarctation of the aorta will have aneurysms of circle of Willis. Their growth appears to be promoted by uncontrolled hypertension. Screening for intracranial aneurysms is not routinely recommended.
remote from the original site of coarctation (201). All patients (repaired and unrepaired) are at risk for endarteritis or endocarditis on an associated bicuspid valve. As many as 10% of patients with coarctation of the aorta will have aneurysms of circle of Willis. Their growth appears to be promoted by uncontrolled hypertension. Screening for intracranial aneurysms is not routinely recommended.
Interruption of the Aortic Arch
Interruption of the aortic arch is defined as the absence of continuity between the transverse arch and the descending thoracic aorta. This uncommon defect produces symptoms in the neonatal period as the ductus arteriosus closes (233




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
