Classification of Cardiomyopathies: Introduction
Cardiomyopathies are an important and heterogeneous group of diseases for which an understanding in both the public and medical community has historically been impaired by confusion surrounding definitions and nomenclature. Classification schemes, of which there have been many,1-8 are useful in defining and drawing relationships or distinctions between these complex diseases for the purpose of promoting greater clarity. Indeed, the precise language of these diseases is profoundly important.
However, many classifications in the literature are to some degree contradictory in design, and indeed none of the proposed schemes can be regarded as ideal (including the most recent and contemporary one presented here). The dilemma is caused by the heterogeneity in the presentation of this diverse group of diseases. A previous prominent classification of cardiomyopathies (1995) was represented in a very brief document under the auspices of the World Health Organization (WHO).1 However, with the identification of new diseases over the past decade, and dramatic advances in cardiovascular diagnosis and knowledge regarding etiology, some disease definitions have become outdated and the WHO classification rendered essentially obsolete. Indeed, the past several years has witnessed a rapid evolution in the molecular genetics of cardiology.9-14 In particular, ion-channelopathies have emerged as conditions predisposing to potentially lethal ventricular tachyarrhythmias, caused by mutations in proteins leading to dysfunctional sodium, potassium, calcium, and other ion channels.
Recently, under the auspices of the American Heart Association, a contemporary classification of cardiomyopathies has been presented,15 relying substantially on recent advances in the characterization of diseases affecting the myocardium.16-18 The new classification scheme affords a large measure of clarity to this area of investigation and facilitates interaction among the clinical and research communities in assessing the diagnosis, prognosis, and management of these complex diseases. This classification takes the place of the WHO document, but as new data emerge it also will undoubtedly require further review and revision.
The contemporary definitions of cardiomyopathies presented here are in concert with the molecular era of cardiovascular disease7 and have direct clinical applications and implications for cardiac diagnosis. However, the classification is a scientific presentation which does not provide methodologies or strategies for clinical diagnosis, nor is it directly applicable to management decisions for patients.
General Considerations
The definition and classification of heart muscle diseases has seen a notable and evolving history. For example, chronic myocarditis was the only recognized cause of heart muscle disease in the 1850s.2 In 1900, the designation of primary myocardial disease was first introduced. However, it was not until 1957 that the term cardiomyopathy was used for the first time. Over the next 25 years, a number of definitions for cardiomyopathies were advanced in concert with an increasing awareness of the nature of these diseases. Indeed, in the original WHO classification,3 cardiomyopathies were defined only as, “heart muscle diseases of unknown cause,” seemingly primitive by today’s standards, and reflecting the paucity of information available regarding etiology and basic disease mechanisms. The most recent WHO definition of cardiomyopathies (1995)1 is, “diseases of myocardium associated with cardiac dysfunction,” and includes newly recognized arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) and primary restrictive cardiomyopathy for the first time and, among unclassified cardiomyopathies, noncompacted left ventricular myocardium.
The diverse cardiomyopathy classifications presented through the years have been designed either for clinicians or biomedical scientists, based on a variety of premises, including etiology, anatomy, physiology, primary treatments, method of diagnosis, biopsy histopathology, and symptomatic state of patients. Although the objective is a classification scheme that can be appreciated by (and can be of use to) all interested parties and disciplines, it is acknowledged that each of the proposed definition and classification constructs have shortcomings. Indeed, it is a reality that no past, present, or future classification of cardiomyopathies is likely to satisfy the purposes of all interested parties.
In particular, the popular hypertrophic-dilated-restrictive cardiomyopathies classification has major limitations by virtue of mixing anatomic designations (ie, hypertrophic and dilated) with a functional one (ie, restrictive). Consequently, confusion frequently arises when the same disease could legitimately appear in two or even three of these categories. Furthermore, such a classification fails to consider the heterogeneous clinical expression and natural history now recognized for many of these diseases. For example, hypertrophic cardiomyopathy (HCM), as well as infiltrative and storage cardiomyopathies are characterized by substantial left ventricular (LV) hypertrophy with increased wall thickness in the absence of ventricular dilatation. These conditions are also frequently associated with restriction to diastolic filling, while purely restrictive forms of cardiomyopathy (without LV hypertrophy) are exceedingly rare. Investigation into the genetic basis of HCM and other cardiomyopathies has led to the identification of individuals with a disease-causing mutation, who nevertheless are without evidence of LV hypertrophy (ie, HCM without hypertrophy). Dilated forms of cardiomyopathy show a considerably increased cardiac mass (ie, weight) with myocyte enlargement indicative of cardiac hypertrophy, although absolute LV wall thicknesses are normal. The end-stage phase of HCM may incorporate hypertrophic and dilated, as well as restrictive components.
Furthermore, some diseases do not have a uniformly static expression and may evolve from one category to another as a consequence of remodeling during their natural clinical course. For example, HCM, amyloid, and other infiltrative myocardial conditions may progress from a nondilated (often hyperdynamic) state with ventricular stiffness to a dilated form with systolic dysfunction and heart failure. Finally, because quantitative assessments of ventricular size represent a continuum and patients can vary widely in the degree of chamber enlargement (and dimensional cutoff values are arbitrary), it is often difficult to discriminate dilated from nondilated forms of cardiomyopathy. This ambiguity may also apply to some rare, or newly identified, cardiac diseases in young patients for which little quantitative cardiac dimensional data are currently available. Indeed, as new cardiomyopathies have been defined in a contemporary fashion (often by genomics), and knowledge of pathologic disease spectrums has evolved, the “dilated-hypertrophic-restrictive” classification19 has become untenable and probably should be abandoned.
Etiologic classifications of cardiomyopathies are also problematic, given that diseases with the same (or similar) phenotypes can harbor diverse etiologies and mechanisms. For example, dilated cardiomyopathy may have genetic, infectious, autoimmune, and toxic causes (and in some cases still designated as idiopathic), all leading however to the final common pathway of ventricular dilatation with systolic dysfunction. Alternatively, functional (ie, physiologic) classifications with potential relevance to treatment considerations and theoretically most useful to clinicians, are also flawed and of limited value since management strategies for these diseases constantly evolve.
Proposed Contemporary Definitions and Classification (2006)
The proposed definition of cardiomyopathies15 is: A heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction, which usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation, and are due to a variety of etiologies that frequently are genetic. Cardiomyopathies are either confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure-related disability.
Within this broad definition, cardiomyopathies are usually associated with failure of myocardial performance, which may be mechanical (eg, diastolic or systolic dysfunction) or as a primary electrical dysfunction prone to life-threatening arrhythmias. The ion channelopathies (long and short QT, Brugada syndromes, and catecholaminergic polymorphic ventricular tachycardia, among others) are primary electrical diseases without gross or histopathologic abnormalities in which the functional and structural myocardial abnormalities responsible for arrhythmogenesis are at the molecular level in the cell or sarcoplasmic membranes themselves. The basic pathologic abnormality in these diseases is not identifiable by either conventional noninvasive imaging or myocardial biopsy during life, or even by electron microscopic or autopsy examination of tissue. The ion-channelopathies are included in this classification of cardiomyopathies based on the scientifically reasonable (but largely hypothetical) assertion that ion channel mutations are responsible for altering biophysical properties and protein structure of the cardiomyocyte, thereby creating structurally abnormal ion channel interfaces and architecture with electrical dysfunction. Consequently, the classification represents a distinct and major departure from prior efforts, and is predicated on the view that causative mutations in genes encoding proteins regulating the transport of ions such as sodium, potassium, and calcium across the cell membranes are ultimately responsible for a structural disease state which triggers primary life-threatening ventricular tachyarrhythmias.
Although the present classification relies substantially on contemporary molecular biology, it is probably premature and inadvisable at this time to formulate a classification entirely dependent on genomics. The molecular genetics of myocardial disease is not yet completely developed and more complex genotype-phenotype relationships will continue to emerge for these diseases. For example, several sarcomeric gene mutations are now known to cause both HCM and dilated cardiomyopathy (DCM). Furthermore, troponin I mutations have been reported to cause both HCM and a purely restrictive form of cardiomyopathy.20
It is also important to specify those disease entities that have not been included as cardiomyopathies in the present contemporary classification. We refer to pathologic myocardial processes which are a direct consequence of other cardiovascular abnormalities that occur with valvular heart disease, systemic hypertension, congenital heart disease, as well as atherosclerotic coronary artery disease (producing ischemic myocardial damage secondary to impaired coronary flow). Therefore, the commonly-used term, ischemic cardiomyopathy, referring to myocardial ischemia and infarction is not relevant to this classification. The following conditions have also been excluded from the cardiomyopathy classification: metastatic and primary intracavitary or intramyocardial cardiac tumors, diseases affecting endocardium with little or no myocardial involvement, as well as the imprecisely defined entity of hypertensive hypertrophic cardiomyopathy.
Cardiomyopathies are divided into two major groups based on predominant organ involvement: Primary cardiomyopathies (genetic, nongenetic, acquired) are those solely or predominantly confined to heart muscle, and are relatively few in number. Secondary cardiomyopathies show pathologic myocardial involvement as part of a large number and variety of generalized systemic (multiorgan) disorders. The systemic diseases associated with secondary forms of cardiomyopathies have previously been referred to as specific cardiomyopathies1 or specific heart muscle diseases3 in prior classifications, but that nomenclature has been abandoned here. The frequency and degree of secondary myocardial involvement varies considerably among cardiomyopathies, some of which are exceedingly uncommon, and for which the evidence of myocardial pathology may be sparse and reported in only a few patients. Because many cardiomyopathies predominantly involve the heart, but are not necessarily confined to that organ, several distinctions between primary and secondary cardiomyopathy made here are necessarily arbitrary, and inevitably rely on judgment regarding the clinical importance and consequences of the myocardial process in patients.
Based on all these considerations, the recommendation15 is that cardiomyopathies can be most effectively classified as (Fig. 31–1):
- Primary-genetic
- Primary-mixed (ie, genetic and nongenetic)
- Primary-acquired
- Secondary
Figure 31–1
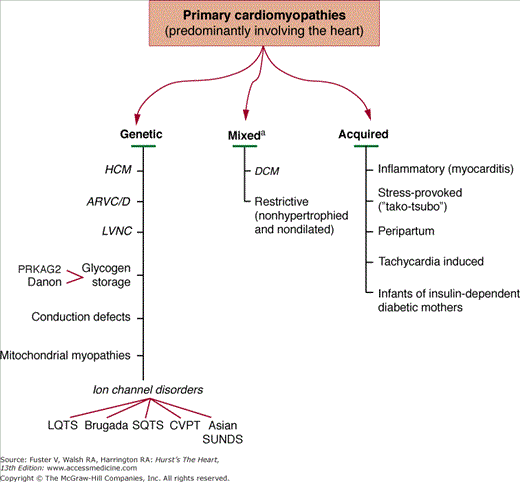
Primary cardiomyopathies in which the clinically relevant disease processes are solely or predominantly confined to the working myocardium. These conditions have been segregated according to their known genetic or nongenetic etiologies.a Familial disease with a genetic etiology reported in a minority of cases. American Heart Association, Inc.15
Primary Cardiomyopathies
HCM is a clinically heterogeneous but relatively common form of genetic heart disease transmitted as an autosomal dominant trait (1:500 of the general population for the disease phenotype recognized by echocardiography), and probably the most frequently occurring cardiomyopathy.9 HCM is the most common cause of sudden cardiac death in the young as well as in trained athletes (in the United States) and is also an important substrate for heart failure disability at any age. Implantable cardioverter-defibrillators (ICDs) have proved lifesaving.21
HCM is characterized morphologically by an otherwise unexplained hypertrophied and nondilated LV in the absence of another cardiac or systemic disease capable of producing the magnitude of wall thickening evident (eg, systemic hypertension, aortic valve stenosis), independent of whether obstruction to LV outflow is present. Histologically, the myocardium is characterized by myocyte disarray, small vessel disease producing microvascular dysfunction, replacement scarring, and increased interstitial fibrosis.22-24 Clinical diagnosis is customarily made with two-dimensional echocardiography (or alternatively with cardiac magnetic resonance [CMR] imaging).25
When LV wall thickness is mild, differential diagnosis with physiologic athlete’s heart may arise. Furthermore, individuals harboring a genetic defect for HCM do not necessarily express clinical markers of their disease at all times during life, such as LV hypertrophy on echocardiogram, ECG abnormalities, or cardiac symptoms. Indeed, ECG alterations can precede the appearance of hypertrophy on echocardiography. Also, virtually any absolute LV wall thickness, even when within normal limits, is consistent with the presence of a HCM-causing mutant gene. On those occasions when hypertrophy is absent, definitive diagnosis can only be made by laboratory-DNA analysis. Furthermore, recognition of LV hypertrophy may be age-related with incomplete penetrance, and initial appearance occasionally delayed in onset well into adulthood (adult morphologic conversion). Most HCM patients have the propensity to develop dynamic obstruction to LV outflow under resting or physiologically provocable (exercise) conditions, produced by systolic anterior motion of the mitral valve and ventricular septal contact.
HCM demonstrates extreme genetic heterogeneity, and is caused by a variety of mutations encoding protein components of the cardiac sarcomere.14 Thirteen mutated sarcomeric genes are presently associated with HCM, most commonly β-myosin heavy chain (the first identified) and myosin-binding protein C. The other genes appear to account for far fewer cases of HCM and include troponin T and I, regulatory and essential myosin light chains, titin, α-tropomyosin, α-actin, α-myosin heavy chain, and muscle LIM protein (MLP). This intergenetic diversity displayed in HCM is compounded by considerable intragenetic heterogeneity, with multiple different mutations identified in each gene (n = >1000 total individual mutations now). These are most commonly missense mutations altering only a single nucleotide (such as with β-myosin heavy chain and α-tropomyosin), although other mutations cause protein truncation (eg, myosin-binding protein C and troponin T). The characteristic diversity of the HCM phenotype is attributable to the disease-causing mutations, but probably also to the influence of modifier genes and environmental factors.
In addition, nonsarcomeric protein mutations in two genes involved in cardiac metabolism have recently been reported to be responsible for primary cardiac glycogen or lysosomal storage cardiomyopathies in older children and adults with a clinical presentation mimicking (or indistinguishable from) that of sarcomeric HCM. One glycogen storage condition involves the gene encoding the γ-2-regulatory subunit of the AMP-activated protein kinase (PRKAG2), associated with variable degrees of LV hypertrophy and ventricular preexcitation.26 A second condition involves the gene encoding lysosome-associated membrane proteins 2 (LAMP-2), resulting in Danon-type storage disease.27 Clinical manifestations are largely limited to the heart, usually with massive degrees of LV hypertrophy and also ventricular preexcitation. These disorders are now part of a subgroup of previously described storage forms of LV hypertrophy such as Pompe disease, a glycogen disease due to alpha-1,4 glycosidase (acid maltase deficiency) in infants and Fabry disease, an X-linked recessive disorder of glycosphingolipid metabolism due to a deficiency of the lysosomal enzyme α-galactosidase A, resulting in intracellular accumulation of glycosphingolipids. The latter disease should be grouped among secondary forms of HCM.
Neither the complete number of HCM disease genes nor the number of HCM-causing mutations is known. Undoubtedly, many other mutations causing cardiac hypertrophy by disrupting sarcomere, metabolic, and other genes remain to be identified. A number of other diseases associated with LV hypertrophy involve prominent thickening of the LV wall, occurring mostly in infants and children ≤4 years of age, which may resemble or mimic typical HCM due to sarcomere protein mutations. These cardiomyopathies also include secondary forms such as Noonan syndrome, an autosomal dominant cardiofacial condition associated with a variety of cardiac defects (most commonly, dysplastic pulmonary valve stenosis and atrial septal defect) due to mutations in PTPN11, a gene encoding the nonreceptor protein tyrosine phosphatase SHP-2 genes.28
Other diseases in this category are mitochondrial myopathies due to mutations encoding mitochondrial DNA (including Kearns-Sayre syndrome), or mitochondrial proteins associated with ATP electron transport chain enzyme defects which alter mitochondrial morphology.29 Also included in these considerations are metabolic myopathies representing ATP production and utilization defects involving abnormalities of fatty acid oxidation (acyl-CoA dehydrogenase deficiencies) and carnitine deficiency, as well as storage myopathies (ie, glycogen storage diseases [type II; autosomal recessive Pompe disease], Hunter and Hurler diseases), and also the transient and nonfamilial cardiomyopathy as part of generalized organomegaly, recognized in infants of insulin-dependent diabetic mothers. In older patients, a number of systemic diseases have been associated with hypertrophic forms of cardiomyopathy; these include Friedreich ataxia, pheochromocytoma, neurofibromatosis, lentiginosis, and tuberous sclerosis, as well as several others.
ARVC/D is an uncommon form of inheritable heart muscle disease (estimated 1:5000), relatively recent in its description only about 20 years ago.30 It is characterized mostly by myocardial electrical instability and risk for life-threatening ventricular arrhythmias. ARVC/D predominantly involves the right ventricle with progressive loss of myocytes and fibrofatty tissue replacement, resulting in regional (segmental) or global abnormalities. Aneurysms of the right ventricle in the triangle of dysplasia (inflow, apex, outflow) are a specific feature.30
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


