Chemokines, Adipokines, and Growth Factors in the Lung
Normal development of organ systems, including the lungs, in utero, during subsequent maturation into adulthood, and in health throughout life, requires intricate signals to be exchanged among various tissues, capable of permitting diversity and adaptation in differing cellular and extracellular contexts, while being robust to onslaughts from ever-changing environmental stimuli. Among the mediators involved in such complex cellular signaling, and relevant in the setting of many pulmonary disorders, are chemokines and growth factors. There has been further complexity added with increasing recognition of adipose tissue as a source of systemic bioactive mediators called adipokines, that impact on lung health. While some of these mediators are implicated in the development of disease states, they are more often than not also critical to tissue homeostasis, and are challenging target systems for therapeutic manipulation. Nonetheless, we are now in an era of exciting developments in targeted biologic therapies that offer the potential for substantial progress in the fight against difficult-to-treat pulmonary disorders characterized by pathogenic processes including acute and chronic inflammation, fibrosis, vascular remodeling, and neoplasia.
CHEMOTACTIC CYTOKINES AND THE INFLAMMATORY RESPONSE
The salient feature of inflammation is leukocyte infiltration. These recruited leukocytes contribute to the pathogenesis of chronic inflammation and promote fibrosis via the elaboration of a variety of cytokines. Maintenance of leukocyte recruitment during inflammation requires the expression of cell surface adhesion molecules, and the production of chemotactic molecules, such as chemokines.1 The chemokines can be divided into four families—CXC, CC, C, and CXXXC—which behave as potent chemotactic factors for neutrophils, eosinophils, basophils, monocytes, mast cells, dendritic cells, NK cells, and T and B lymphocytes (Table 26-1). There is approximately 20% to 40% homology between the members of the four chemokine families.2 Chemokines are produced by an array of cells, including monocytes, alveolar macrophages, neutrophils, platelets, eosinophils, mast cells, T and B lymphocytes, NK cells, and various structural cells, including keratinocytes, mesangial cells, epithelial cells, hepatocytes, fibroblasts, smooth muscle cells, mesothelial cells, and endothelial cells. Production of chemokines by both immune and nonimmune cells supports the contention that these cytokines may play a pivotal role in orchestrating chronic inflammation.3
TABLE 26-1 The Human C, CC, CXC, and CXXXC Chemokine Families of Chemotactic Cytokines

 CXC CHEMOKINES
CXC CHEMOKINES
CXC chemokines can be further divided into two groups on the basis of a structure/function domain consisting of the presence or absence of three amino acid residues (Glu-LeuArg; ELR motif) that precede the first cysteine amino acid residue in the primary structure of these cytokines. ELR+CXC chemokines are chemoattractants for neutrophils and act as potent angiogenic factors. In contrast, ELR–CXC chemokines are highly induced by interferons, are chemoattractants for mononuclear cells, and are potent inhibitors of angiogenesis (Table 26-2).4
TABLE 26-2 The CXC Chemokines that Display Disparate Angiogenic Activity
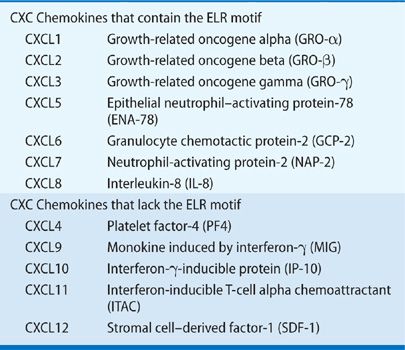
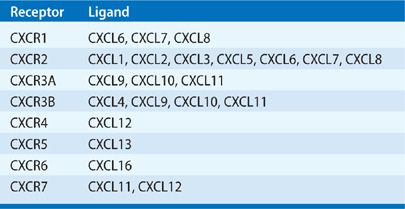
Chemokine activities are mediated through G-protein–coupled receptors. Seven CXC chemokine receptors have been identified (Table 26-3). The ELR+ chemokines bind to CXCR1 and CXCR2 receptors, which are found on neutrophils, T lymphocytes, monocytes/macrophages, eosinophils, basophils, keratinocytes and mast cells, and endothelial cells.5 CXCR3 is the receptor for CXCL9, CXCL10, and CXCL11, and is expressed on activated T lymphocytes. CXCR3 is also expressed on human umbilical vein endothelial cells (HUMVECs) in a cell cycle–dependent fashion. CXCR4 is the specific receptor for CXCL12 and is the cofactor for lymphotropic HIV-1. In contrast to CXCR3, CXCR4 appears to be expressed on resting T lymphocytes.5 Two other chemokine receptors have been identified that bind chemokines without a subsequent signal-coupling event. The DARC receptor is similar to other chemokine receptors and it binds both CXC and CC chemokines without apparent signal coupling. This receptor was originally found on human erythrocytes and was thought to represent a “sink” for chemokines.6 The second nonsignaling chemokine receptor is the D6 receptor, which binds several CC chemokines with high affinity, including CCL2, CCL4, CCL5, and CCL7.7
TABLE 26-3 The CXC Chemokine Receptors
CXC Chemokines in Pulmonary Inflammation
CXC chemokines play a significant role in mediating neutrophil infiltration in the lung parenchyma and pleural space in response to endotoxin and bacterial challenge. CXCL8 is in the bronchoalveolar lavage of patients with community-acquired pneumonia and nosocomial pneumonia and a variety of animal models of pneumonia. In a model of Aspergillus fumigatus pneumonia, neutralization of TNF resulted in marked attenuation of the expression of CXCL1 and CXCL2/3 that was paralleled by a reduction in the infiltration of neutrophils and associated with increased mortality.8 Administration of a TNF agonist peptide to animals that had been intratracheally inoculated with Klebsiella pneumoniae led to markedly elevated levels of CXCL2/3 associated with increased neutrophil infiltration.9 Studies have shown that ventilator-induced lung injury is secondary to stretch-induced chemokine release with a subsequent inflammatory response and neutrophil recruitment.10 CXCR2 -/- mice are also protected from hyperoxia-induced lung injury. In other studies, the production of CXCL5 in the lung was correlated with the presence of neutrophil-dependent lung injury, and passive immunization with neutralizing CXCL5 antibodies resulted in significant attenuation of lung injury.11
Several studies have demonstrated that CXCL8 levels correlate with the development and mortality of ARDS. Early increases in CXCL8 in bronchoalveolar lavage fluid correlated with an increased risk of subsequent development of ARDS, and also demonstrated that alveolar macrophages were an important source of CXCL8 prior to neutrophil influx.10 Furthermore, there is an imbalance in the expression of ELR+ (CXCL1, CXCL5, CXCL8) as compared with ELR–CXC (CXCL10, CXCL11) chemokines from bronchoalveolar lavage fluid (BALF) of patients with ARDS as compared with controls. This imbalance correlated with angiogenic activity and both procollagen I and procollagen III levels in BALF.12 These findings suggest that CXC chemokines have an important role in the fibroproliferative phase of ARDS via the regulation of angiogenesis.
The Role of CXC Chemokines in Pulmonary Fibrosis
IPF is characterized by the progressive deposition of collagen within the interstitium and subsequent destruction of lung tissue.13,14 The mechanisms of cellular injury and the role of classic inflammatory cells remain unclear. CXCL8 is significantly elevated in IPF, as compared with either normal or sarcoidosis patients, and correlates with BALF presence of neutrophils. The alveolar macrophage is an important cellular source of CXCL8 in IPF.15 In addition, BALF levels of CXCL8 in IPF may correlate with a worse prognosis.16
Vascular Remodeling in Pulmonary Fibrosis: The Role of CXC Chemokines
The existence of neovascularization in IPF was originally identified in 1963 by Turner-Warwick, who demonstrated that within areas of pulmonary fibrosis there was extensive neovascularization with anastomoses between the systemic and pulmonary microvasculature.17 Further evidence of neovascularization during the pathogenesis of pulmonary fibrosis has been demonstrated in a rat model of bleomycin-induced pulmonary fibrosis.18 An imbalance in the levels of angiogenic chemokines (CXCL5, CXCL8), as compared with angiostatic chemokines (CXCL9, CXCL10, CXCL11), favoring net angiogenesis has been demonstrated in both animal models and tissue specimens from patients with IPF (Fig. 26-1).19 Renzoni20 has demonstrated vascular remodeling in both IPF and fibrosing alveolitis associated with systemic sclerosis. Cosgrove et al.21 provided further support for the concept of vascular remodeling in IPF when they demonstrated a relative absence of vessels in the fibroblastic foci of IPF. This appeared to correlate with increased expression of pigment epithelium–derived factor in the fibroblastic foci. Interestingly, they also noted significant vascularity in the areas of fibrosis around the fibroblastic foci, with numerous abnormal vessels in the regions of severe architectural distortion. These findings are similar to those of Renzoni and support the concept of regional heterogeneity of vascularity in IPF. This heterogeneity is not surprising, as usual interstitial pneumonia, which is the pathologic description of IPF, is defined by its regional and temporal heterogeneity.14
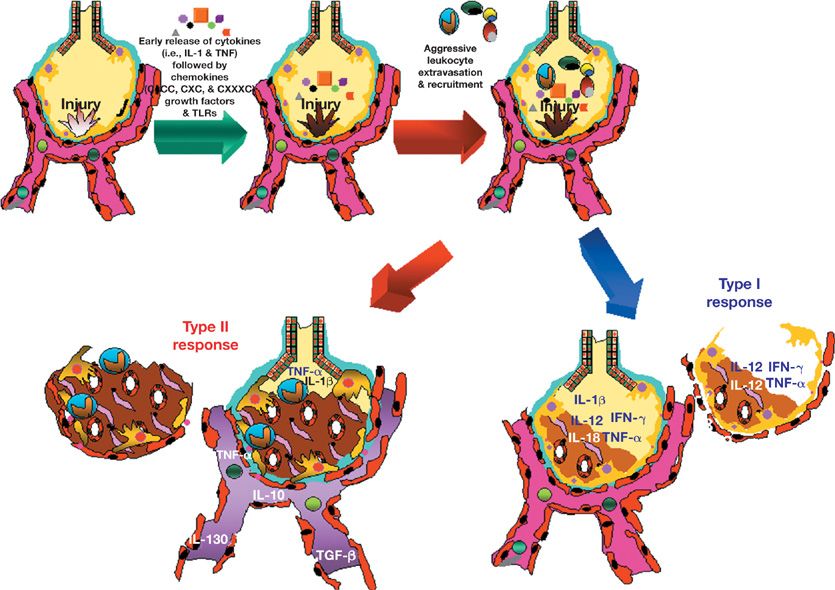
Figure 26-1 The inflammatory response to lung injury. The cytokine profile that is secreted by inflammatory cells during lung injury determines the ultimate outcome following injury. Polarization of the inflammatory response toward a type I response is associated with resolution of lung injury or infection. In contrast chronic infections (e.g., tuberculosis) and chronic inflammatory diseases (e.g., idiopathic pulmonary fibrosis) are associated with a type II profile.
CXC Chemokines in Pulmonary Hypertension
The potential role of the CXCL12/CXCR4/CXCR7 axis in pulmonary hypertension was first suspected when it was reported that CXCR7 expression increased in the lungs of hypoxic hypertensive mice and in the lungs of patients with idiopathic pulmonary arterial hypertension (IPAH).22 Subsequently, CXCL12 was found to be elevated in the peripheral plasma of patients with pulmonary artery hypertension (PAH), although in a separate study this increase was not observed.23,24 Increased expression of CXCL12 in remodeled vessels and particularly in the plexiform lesions in explanted IPAH lungs has also been shown.25
CXR7 was most prominently expressed in the endothelium of the explanted lungs of hypertensive subjects and, in addition to its well-established role in leukocyte chemotaxis, was shown to play a central role in stimulating endothelial proliferation, whereas CXCR4 was required for endothelial cell chemotaxis.23 Recruitment of progenitor cells to the remodeled pulmonary vessels is a prominent feature in PAH lungs.25,26 In vivo blockade of CXCR4 in hypoxic mice reduced the recruitment of progenitor cells to the remodeled vasculature of hypertensive lungs, whereas blockade of CXCR7 did not affect this behavior.24,26,27
In rodent models inhibition of CXCL12 action using function-blocking antibodies, or separate selective inhibition of CXCR4 signaling or CXCR7 signaling, attenuates the development of hypoxic pulmonary hypertension.24,27,28 Taken together these data suggest an important role of CXCL12 signaling, requiring both its cognate receptors, in the pathogenesis of pulmonary hypertension.
 CHEMOKINES AND THE TRAFFICKING OF FIBROCYTES TO THE LUNG
CHEMOKINES AND THE TRAFFICKING OF FIBROCYTES TO THE LUNG
Fibrocytes present in the peripheral circulation were first identified in 1994, comprise a minor component of the circulating pool of leukocytes (less than 1%), and express a characteristic pattern of markers, including collagen (Col) I and CD45.29 Subsequent studies have revealed that circulating fibrocytes express chemokine receptors such as CXCR4 and CCR7 and extracellular matrix (ECM) proteins, such as procollagen I and procollagen III. Fibrocytes migrate in response to CXCL12 and traffic to the lungs in a model of bleomycin-induced pulmonary fibrosis. Treatment of bleomycin-exposed animals with specific neutralizing anti-CXCL12 antibodies inhibits intrapulmonary recruitment of fibrocytes and attenuated lung fibrosis.30 Levels of circulating CD45+ Col1+ cells are elevated in stable IPF patients versus controls, are transiently higher during an exacerbation and predicted survival.31 These findings challenge the dogma that fibroblasts and myofibroblasts arise from an intrapulmonary pool of tissue fibroblasts.32
 THE CC CHEMOKINES
THE CC CHEMOKINES
CC chemokines (Table 26-1) are chemoattractants for monocyte, T and B lymphocytes, NK cells, dendritic cells, basophils, mast cells, and eosinophils.33 The CC chemokines are produced by an array of cells, including monocytes, alveolar macrophages, neutrophils, platelets, eosinophils, mast cells, T cells, B cells, and NK cells, as well as structural cells such as keratinocytes, mesangial cells, epithelial cells, hepatocytes, fibroblasts, smooth muscle cells, mesothelial cells, and endothelial cells.34
CC Chemokine Receptors
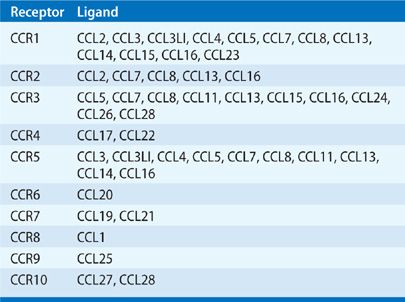
CC chemokine receptors are structurally homologous and have been identified to have specific ligand-binding profiles (Table 26-4).34 Naïve T cells express CXCR4 and CCR7 and migrate in response to CXCL12 and CCL19. CXCR3, CXCR6, and CCR5 are expressed at higher levels on type I cells than type II, whereas CCR3, CCR4, and CCR8 are more characteristic of type II cells.35
TABLE 26-4 The CC Chemokine Receptors
CC Chemokines in Pulmonary Inflammation
The CC chemokines, CCL2, CCL3, CCL4, CCL5 have been implicated in mediating the innate host defense in animal models of pulmonary infection.36,37 These studies have demonstrated that CC chemokine ligand/receptor biology plays a critical role in innate host defense and development of pulmonary inflammation that is important in eradication of microorganisms.
Mehrad et al. have shown that CCL3 and the recruitment of mononuclear cells play an important role in the eradication of invasive pulmonary aspergillosis. They demonstrated that in both immunocompetent and neutropenic mice CCL3 is induced in the lungs in response to intratracheal inoculation of A. fumigatus.38 These studies indicate that CCL3 and elicitation of mononuclear cells are crucial in mediating host defense against A. fumigatus in the setting of neutropenia.39
CC Chemokines in Pulmonary Fibrosis
Animal models, such as bleomycin-induced pulmonary fibrosis, have demonstrated the presence and contribution of CC chemokines to the pathogenesis of fibrosis. CCL2 is an important cofactor for the stimulation of fibroblast collagen production and induction of the expression of TGF-β1. Inhibition of CCL2 or CCL3 resulted in a reduction of infiltrating cells into the lungs of bleomycin-treated animals.40,41
Furthermore, it has been shown that CCL2 can stimulate interleukin-4 (IL-4) production, indicating that it might be involved in type II polarization.42 IL-13 promotes bleomycin-induced fibrosis through the elaboration of CCL6.43 Both CCL17 and CCL22 and their receptor, CCR4, are significantly elevated in the bleomycin model and neutralization of CCL17 attenuates pulmonary fibrosis.44 Thus, chemokines may have an important role in the switch toward a profibrotic type II phenotype.
Both CCR1 and CCR2 have been shown to play an important role in the pathogenesis in the mouse model of bleomycin-induced pulmonary fibrosis. Treatment with antibodies to CCR1 leads to a reduction in both inflammatory cell infiltrates and the development of fibrosis.45 Similarly, CCR2 -/- mice are protected from pulmonary fibrosis in response to bleomycin.46 Furthermore, alveolar epithelial cells from CCR2 -/- mice suppress fibroblast proliferation more than AECs from wild-type mice.47 CCL2 and CCR2 have an important role in suppression of PGE2, thereby promoting fibroproliferation. Similarly, an important role for CCR2 has been seen in murine model of obliterative bronchiolitis, in which the fibrotic response associated with this disorder was attenuated in CCR2 -/- mice.48 Similarly, CCL2 and CCL3 are elevated in BALF and lung tissue of ILD patients.49,50 Accordingly, targeting chemokine receptors may be an efficient way to inhibit pulmonary fibrosis.
Choi et al.51 described enhanced expression of the chemokines CCL7 and CCL22, in lung tissue of patients with IPF as compared with nonspecific interstitial pneumonia, and nonidiopathic interstitial pneumonia. Furthermore, they describe increased expression of CCL5 in nonspecific interstitial pneumonia as compared with usual interstitial pneumonia. Interestingly, CCL5 protein was identified in nonspecific interstitial pneumonia more prominently than usual interstitial pneumonia. This is all the more interesting as CCL5 is a major stimulus for the production of CCL7 through its interactions with CCR5. These findings raise the possibility that there is a continuum from nonspecific interstitial pneumonia to usual interstitial pneumonia with higher levels of CCL5 in nonspecific interstitial pneumonia leading to subsequent increased CCL7 expression as the disease progresses to usual interstitial pneumonia. There is considerable controversy as to whether nonspecific interstitial pneumonia is an earlier lesion of usual interstitial pneumonia.52 Several studies have demonstrated the presence of usual interstitial pneumonia and nonspecific interstitial pneumonia patterns in the same patients, which suggests that these are overlapping processes.52–54 The findings of Choi suggest a transition from a predominance of CCL5 to CCL7 and further support this notion. Of further interest is the previous description that CCL7 can act as a natural antagonist at the CCR5 receptor, which raises the possibility that CCL7 may play a role in regulating its own production.55
ADIPOKINES AND PULMONARY INFLAMMATION
With the emergence, in the 20th century, of obesity as a major epidemic linked to systemic metabolic dysfunction, concerted efforts have been made to gain a fuller understanding of the varied functions of adipose tissue, beyond that of being an energy storage organ, and leading to the discovery of various factors that are secreted by adipocytes.56 These mainly proteinaceous endocrine factors were initially termed adipocytokines, and later, adipokines,57 and possess pro- and anti-inflammatory activities, potentially of relevance in the context of pulmonary disorders associated with body mass index, including asthma, obstructive sleep apnea syndrome, chronic obstructive pulmonary disease (COPD), PAH, various pulmonary infections, and lung cancer.58 While the main source of adipocytes in the body is from deposits of subcutaneous and visceral adipose tissue, the development of obesity can give rise to collections of adipose tissue in other locations including the heart, kidneys, bone marrow, the adventitia of major blood vessels, and within the lungs. There is evidence that differential adipokine secretion and functional outcomes can occur at different sites of adiposity within the body, following the stimulus of dietary modification.59 White adipose tissue, the major form in humans, is predominantly composed of lipid-laden adipocytes, but also adipocyte precursor cells, fibroblasts that generate ECM scaffolding, vascular (smooth muscle and endothelial) cells that provide systemic access for secreted adipokines, and macrophages and T cells that influence the immune phenotype.60 The cellular composition of adipose tissue can vary in conditions of altered body mass, and macrophages in particular, seem to traffic in larger numbers to adipose tissue under conditions of increased obesity with associated capillary rarefaction and adipose tissue hypoxia.61
 PROINFLAMMATORY ADIPOKINES
PROINFLAMMATORY ADIPOKINES
Beyond the “pure” effects of uncomplicated obesity upon respiratory mechanics, pulmonary gas exchange, ventilatory drive, and work of breathing, there is increasing evidence of an association among obesity, adipokines, and pulmonary disease states that are characterized by inflammation.58 Most of the adipokines identified to date are proinflammatory in their effects (Table 26-5). Some important adipokines that are better known for their other roles, such as TNF-α, IL-6, CCL2, and CXCL5, will not be discussed here.
TABLE 26-5 Adipokines Linked to Pulmonary Disease States
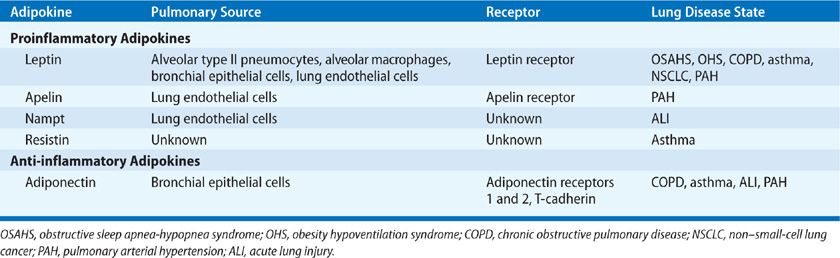
Leptin
Leptin, coded for by the ob gene on chromosome 17, is a 16 KDa protein hormone, mainly secreted by adipocytes,62 and regulated principally by food intake, whereby fasting reduces leptin levels, and food consumption transiently increases ob gene expression, with leptin being initially regarded as a satiety hormone.63,64 It is also expressed in human peripheral lung tissue, including alveolar type II pneumocytes, alveolar macrophages, and bronchial epithelial cells,65,66 and is known to be modulated by gender, sepsis, catecholamines, glucocorticoids, and insulin.67,68 It is now clear that leptin has pleiotropic effects, which include stimulating TNF and IL-6 production by monocytes, stimulating production of CCL3–5 by macrophages, and stimulating reactive oxygen species production, cell proliferation, and migration. In addition, leptin may have a role in lung development,56 as it is differentially expressed by fetal rat lung fibroblasts during the time of alveolar differentiation,69 and capable of stimulating fetal rat surfactant protein synthesis in vitro,70 although contradictory in vivo findings were observed in fetal sheep and mice lungs.71
There have been extensive efforts to relate leptin to respiratory disorders, most notably with obstructive sleep apnea-hypopnea syndrome (OSAHS), COPD, asthma, pneumonia, and other pulmonary infections. Some data suggest leptin has a stimulatory effect on ventilation; for example, the mutant ob/ob mouse, which lacks functional leptin and has a phenotype of obesity, hyperphagia, and a low-resting metabolic rate, displays an elevated arterial PaCO2 independent of obesity onset, which is acutely reversible through exogenous leptin replacement.72 It has been speculated that OSAHS is a leptin-resistant state, but progress in this field has been hampered by conflicting results of leptin and leptin receptor candidate gene association studies73,74 and trials of nasal continuous positive airway pressure.75,76
In COPD, it has been hypothesized that the known link between the cachexia of COPD and increased mortality, might somehow be related to leptin function. There appears to be an absence of the usual circadian rhythm of circulating leptin in cachectic COPD patients, as opposed to normal weight COPD patients, with associated heart rate variability changes that raise the possibility of a role for leptin in the pathophysiology of COPD cachexia,77 although further data are needed. The reproducible finding of elevated serum leptin levels during acute exacerbations of COPD, that track markers of the systemic inflammatory response, points to disturbance of the normal leptin feedback loop regulating food intake and energy balance, due to the influence of the acute inflammatory response and systemically administered glucocorticoids, as a possible contributing mechanism to cachexia in COPD.78 Wild-type mice exposed chronically to cigarette smoke have increased leptin expression in bronchial epithelial cells and pneumocytes versus air-exposed controls. Ob/ob mice (lacking functional leptin) that are then exposed acutely or chronically to cigarette smoke exhibit higher neutrophils, CD4+, CD8+, and dendritic cells in BAL and lung tissue than smoke-exposed wild-type mice, compatible with modulation of innate and adaptive immune cell recruitment by leptin in response to smoke.79 Within human airways, leptin expression is increased in bronchial epithelial cells and alveolar macrophages of ex-smokers with or without severe COPD compared to never smokers, and leptin can induce phosphorylation of the transcription factor STAT3 in lung epithelial cells, supporting the notion of a functioning leptin signaling pathway in these cells.65
Obesity is believed to be a risk factor for asthma, but some controversy still surrounds whether or not obesity is a central cause or a confounding comorbidity of asthma.80,81 Notably, unbiased clustering approaches in severe asthma subjects have identified an obesity-related clinical asthma phenotype typified by obese, female patients with late-onset asthma, and lacking in eosinophilic Th2-mediated inflammation.82 Ob/ob mice, a model of loss of leptin function, appear to have elevated pulmonary resistance, and increased responses to ozone and methacholine, though this could be mechanical bias from the low lung size of these mutant mice.68 Interestingly, when a cohort of obese asthmatic women and obese female controls were studied in the setting of bariatric surgery, leptin expression and macrophage inflammation were both increased in visceral adipose tissue of asthmatics independent of BMI, and correlated with airway reactivity, but there was no association with airway inflammation measurements even though the airway epithelial cells expressed receptors for leptin, suggesting that leptin exerts a direct effect on airway epithelium (and not indirectly via enhancing airway inflammation) that is important in the pathogenesis of asthma in obesity.83 These observations run counter to the hypothesis that leptin is merely a marker for airway inflammation in poorly controlled asthma.84
Nampt (PBEF/visfatin)
The gene Nampt codes for a cytokine called pre–B cell colony-enhancing factor (PBEF), also known as visfatin. It is now known to be produced by various cell types including adipocytes, and especially by visceral as opposed to subcutaneous fat, and has gained greater interest by the rediscovery of PBEF as Nampt, the rate-limiting enzyme in the biosynthesis of the essential redox cofactor, NAD. Nampt is inducible in neutrophils and lung microvascular endothelial cells by endotoxin, TNF-α and IL-1β, and in monocytes, it induces production of IL-1β, TNF-α, and IL-6, and the surface expression of costimulatory molecules CD54, CD40, and CD80.85,86 Against this background of catalyzing the respiratory burst, and its proinflammatory and immunomodulatory capabilities, Nampt has been demonstrated to be found at higher concentrations in serum and BAL fluid from patients with acute lung injury.87 There have also been a number of independent candidate gene association studies that support a role for Nampt promoter genetic variants in ALI pathogenesis.87–89
 ANTI-INFLAMMATORY ADIPOKINES
ANTI-INFLAMMATORY ADIPOKINES
Adiponectin
The most intensely studied of the anti-inflammatory adipokines, and with notable evidence of a role in inflammatory lung disorders, is adiponectin, which shares some structural similarity with complement factor C1q, and forms trimers that can go on to form stable hexamers or a high molecular weight form, all of which are detectable in blood.60 Circulating adiponectin levels are decreased in obesity, particularly in visceral obesity, nonalcoholic fatty liver disease, and type 2 diabetes, being inversely related to insulin resistance. The adipokine is inhibited by proinflammatory factors such as TNF, IL-6, hypoxia, and oxidative stress.56 There has been considerable interest concerning the involvement of adiponectin in pulmonary disease states. Three described adiponectin receptors (AdipoR1, AdipoR2, and T-cadherin) are expressed in the lungs, with AdipoR1 expressed by lung epithelial cells, and importantly, the baseline phenotype of mice following targeted disruption of the adiponectin gene shows evidence of emphysema-like dilated air spaces and activation of alveolar macrophages.90,91 These same adiponectin-deficient mice develop pulmonary hypertension with evidence of perivascular inflammation,92 and an exaggerated form of LPS-inducible acute lung injury that is abrogated by adiponectin.93
As further evidence of anti-inflammatory functionality, adiponectin can bind to apoptotic cells and facilitate their phagocytosis by macrophages, analogous to other members of the collectin family, such as C1q and surfactant proteins A and D.94 In keeping with this intuitive role in airways disease pathobiology, a number of groups have reported increased levels of circulating adiponectin in COPD.91,95 Adiponectin was found to be elevated in BALF of COPD patients, being highly expressed in their airway epithelium, and the adiponectin receptor AdipoR1 on lung epithelial cells was shown to be functional, releasing IL-8 in the presence of adiponectin.91 Animal and human data suggest that short-term exposure to cigarette smoke downregulates adiponectin whereas the COPD disease state elevates its expression.91 In contrast, AdipoR2 is more highly expressed in the airway epithelium of obese asthma patients versus obese control subjects, and the opposite to what is observed for T-cadherin expression.83 In mice, allergen challenge appears to decrease the pulmonary expression of all three adiponectin receptor types, and T-cadherin may play a role in transporting adiponectin into the lungs.96 The exact role of adiponectin signaling in asthma will require further study.
 ADIPOKINES AND PULMONARY HYPERTENSION
ADIPOKINES AND PULMONARY HYPERTENSION
The high prevalence of pulmonary hypertension in obese subjects has previously been attributed to hypoxemia resulting from associated obstructive sleep apnea or obesity hypoventilation syndrome.97–99 However, more recently evidence has been presented that alterations in three specific adipokines (adiponectin, apelin, and leptin) may directly contribute to pulmonary vascular dysfunction.
Adiponectin concentrations are paradoxically reduced in the plasma of obese subjects despite the expansion of adipose tissue.100 Mice in which adiponectin has been deleted spontaneously develop elevated pulmonary arterial pressure, vascular remodeling, and perivascular inflammation as they grow older.92 Conversely constitutive overexpression of adiponectin in mice protects against the development of both hypoxia-induced and inflammation-induced (ovalbumin challenge) pulmonary hypertension.101 These suggest that reduced adiponectin concentrations contribute directly to the development of pulmonary vascular dysfunction in obesity due to a loss of its normal vascular homeostatic action.
Apelin is produced and secreted by adipocytes and acts by binding to the apelin receptor.102 It is also expressed by a wide variety of other tissues including the pulmonary vasculature.102 Plasma apelin concentrations are increased in obese individuals.103 Apelin null mice develop more severe hypoxic pulmonary hypertension than wild-type mice, which is associated with endothelial dysfunction.104 Leptin concentrations are elevated in patients with both idiopathic PAH and scleroderma-associated PAH and in the pulmonary endothelium from these patients.105,106 However, the exact role of apelin and leptin in the pulmonary circulation in obese humans remains to be determined.
GROWTH FACTORS
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


