CARDIOGENIC SHOCK AND PULMONARY EDEMA
Judith S. Hochman ![]() David H. Ingbar
David H. Ingbar
Cardiogenic shock and pulmonary edema are life-threatening conditions that should be treated as medical emergencies. The most common etiology for both is severe left ventricular (LV) dysfunction that leads to pulmonary congestion and/or systemic hypoperfusion (Fig. 28-1). The pathophysiology of pulmonary edema is discussed in Chap. 5.
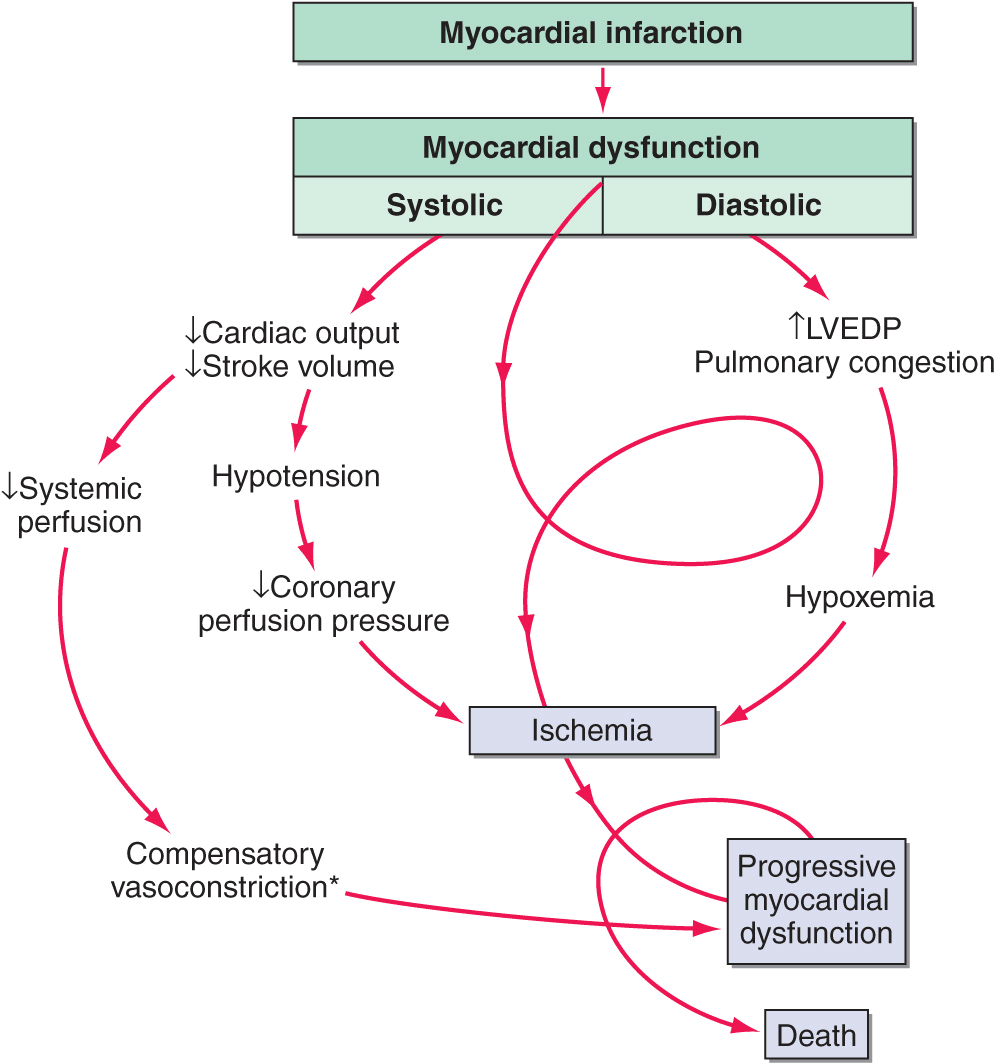
FIGURE 28-1
Pathophysiology of cardiogenic shock. Systolic and diastolic myocardial dysfunction results in a reduction in cardiac output and often pulmonary congestion. Systemic and coronary hypoperfusion occur, resulting in progressive ischemia. Although a number of compensatory mechanisms are activated in an attempt to support the circulation, these compensatory mechanisms may become maladaptive and produce a worsening of hemodynamics. *Release of inflammatory cytokines after myocardial infarction may lead to inducible nitric oxide expression, excess nitric oxide, and inappropriate vasodilation. This causes further reduction in systemic and coronary perfusion. A vicious spiral of progressive myocardial dysfunction occurs that ultimately results in death if it is not interrupted. LVEDP, left ventricular end-diastolic pressure. (From SM Hollenberg et al: Ann Intern Med 131:47, 1999.)
CARDIOGENIC SHOCK
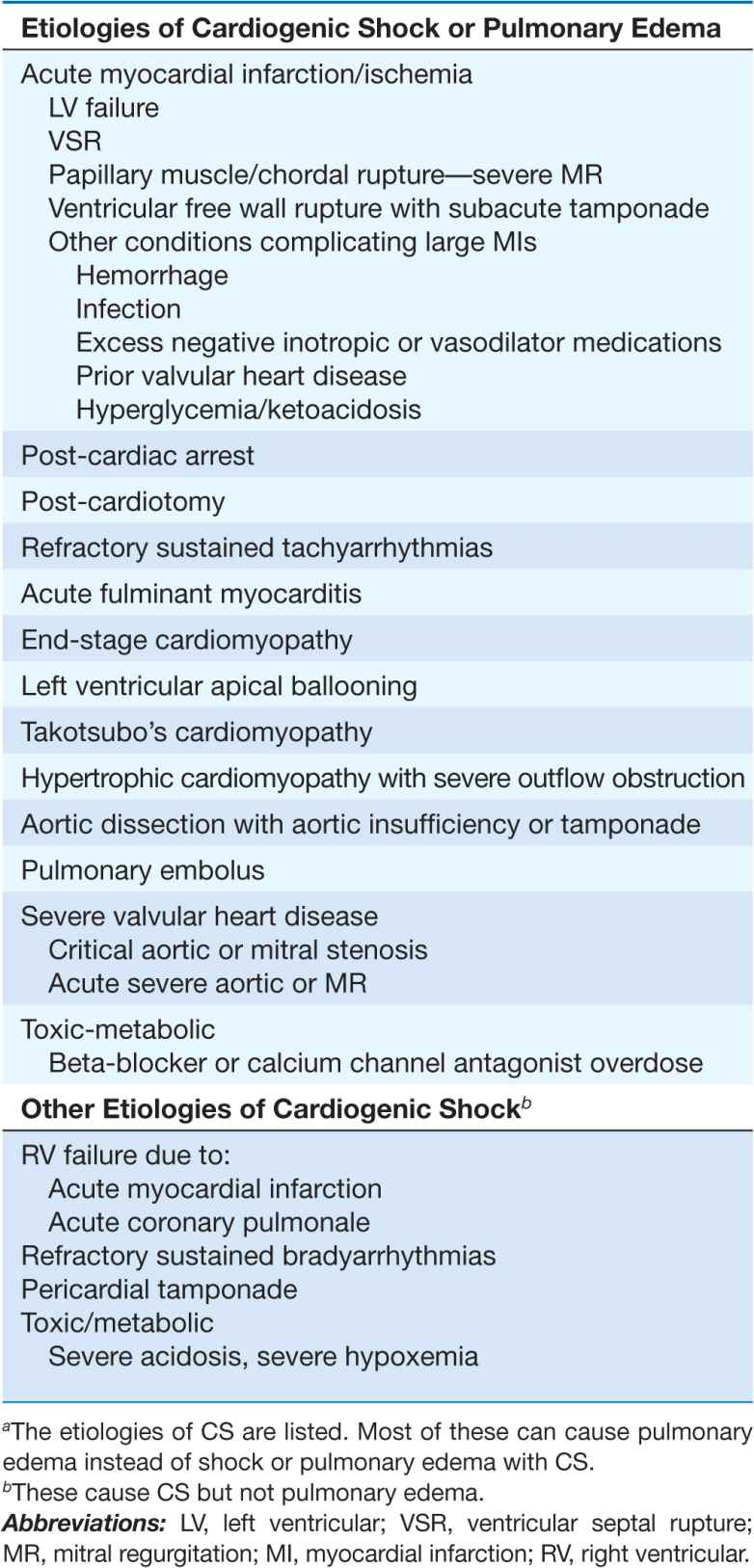
Cardiogenic shock (CS) is characterized by systemic hypoperfusion due to severe depression of the cardiac index (<2.2 [L/min]/m2) and sustained systolic arterial hypotension (<90 mmHg) despite an elevated filling pressure (pulmonary capillary wedge pressure [PCWP] >18 mmHg). It is associated with in-hospital mortality rates >50%. The major causes of CS are listed in Table 28-1. Circulatory failure based on cardiac dysfunction may be caused by primary myocardial failure, most commonly secondary to acute myocardial infarction (MI) (Chap. 35), and less frequently by cardiomyopathy or myocarditis (Chap. 21), cardiac tamponade (Chap. 22), or critical valvular heart disease (Chap. 20).
TABLE 28-1
ETIOLOGIES OF CARDIOGENIC SHOCK (CS)a AND CARDIOGENIC PULMONARY EDEMA
Incidence
CS is the leading cause of death of patients hospitalized with MI. Early reperfusion therapy for acute MI decreases the incidence of CS. The rate of CS complicating acute MI was 20% in the 1960s, stayed at ~8% for >20 years, but decreased to 5–7% in the first decade of this millennium. Shock typically is associated with ST elevation MI (STEMI) and is less common with non-ST elevation MI (Chap. 35).
LV failure accounts for ~80% of cases of CS complicating acute MI. Acute severe mitral regurgitation (MR), ventricular septal rupture (VSR), predominant right ventricular (RV) failure, and free wall rupture or tamponade account for the remainder.
Pathophysiology
CS is characterized by a vicious circle in which depression of myocardial contractility, usually due to ischemia, results in reduced cardiac output and arterial pressure (BP), which result in hypoperfusion of the myocardium and further ischemia and depression of cardiac output (Fig. 28-1). Systolic myocardial dysfunction reduces stroke volume and, together with diastolic dysfunction, leads to elevated LV end-diastolic pressure and PCWP as well as to pulmonary congestion. Reduced coronary perfusion leads to worsening ischemia and progressive myocardial dysfunction and a rapid downward spiral, which, if uninterrupted, is often fatal. A systemic inflammatory response syndrome may accompany large infarctions and shock. Inflammatory cytokines, inducible nitric oxide synthase, and excess nitric oxide and peroxynitrite may contribute to the genesis of CS as they do to that of other forms of shock. Lactic acidosis from poor tissue perfusion and hypoxemia from pulmonary edema may result from pump failure and then contribute to the vicious circle by worsening myocardial ischemia and hypotension. Severe acidosis (pH <7.25) reduces the efficacy of endogenous and exogenously administered catecholamines. Refractory sustained ventricular or atrial tachyarrhythmias can cause or exacerbate CS.
Patient profile
In patients with acute MI, older age, female sex, prior MI, diabetes, and anterior MI location are all associated with an increased risk of CS. Shock associated with a first inferior MI should prompt a search for a mechanical cause. Reinfarction soon after MI increases the risk of CS. Two-thirds of patients with CS have flow-limiting stenoses in all three major coronary arteries, and 20% have stenosis of the left main coronary artery. CS may rarely occur in the absence of significant stenosis, as seen in LV apical ballooning/Takotsubo’s cardiomyopathy.
Timing
Shock is present on admission in only one-quarter of patients who develop CS complicating MI; one-quarter develop it rapidly thereafter, within 6 h of MI onset. Another quarter develop shock later on the first day. Subsequent onset of CS may be due to reinfarction, marked infarct expansion, or a mechanical complication.
Diagnosis
Due to the unstable condition of these patients, supportive therapy must be initiated simultaneously with diagnostic evaluation (Fig. 28-2). A focused history and physical examination should be performed, blood specimens sent to the laboratory, and an electrocardiogram (ECG) and chest x-ray obtained.
Echocardiography is an invaluable diagnostic tool in patients suspected of CS.
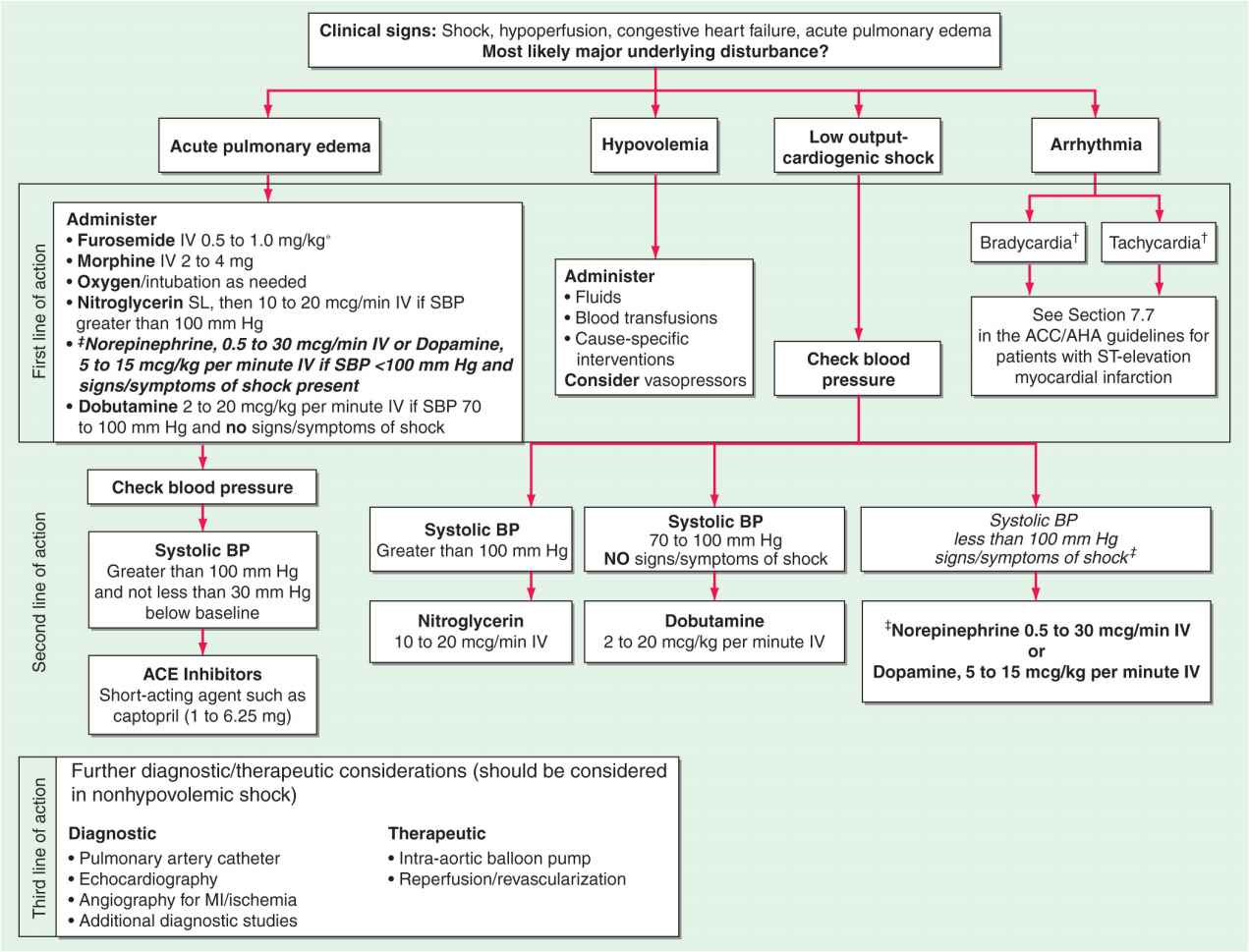
FIGURE 28-2
The emergency management of patients with cardiogenic shock, acute pulmonary edema, or both is outlined. *Furosemide: <0.5 mg/kg for new-onset acute pulmonary edema without hypervolemia; 1 mg/kg for acute on chronic volume overload, renal insufficiency.†For management of bradycardia and tachycardia, see Chaps. 15 and 16.‡Indicates modification from published guidelines. ACE, angiotensin-converting enzyme; BP, blood pressure; MI, myocardial infarction. (Modified from Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 7: The era of reperfusion: Section 1: Acute coronary syndromes [acute myocardial infarction]. The American Heart Association in collaboration with the International Liaison Committee on Resuscitation. Circulation 102:I172, 2000.)
 Clinical findings
Clinical findings
Most patients have continuing chest pain and dyspnea and appear pale, apprehensive, and diaphoretic. Mentation may be altered, with somnolence, confusion, and agitation. The pulse is typically weak and rapid, often in the range of 90–110 beats/min, or severe bradycardia due to high-grade heart block may be present. Systolic blood pressure is reduced (<90 mmHg) with a narrow pulse pressure (<30 mmHg), but occasionally BP may be maintained by very high systemic vascular resistance. Tachypnea, Cheyne-Stokes respirations, and jugular venous distention may be present. The precordium is typically quiet, with a weak apical pulse. S1 is usually soft, and an S3 gallop may be audible. Acute, severe MR and VSR usually are associated with characteristic systolic murmurs (Chap. 35). Rales are audible in most patients with LV failure causing CS. Oliguria (urine output <30 mL/h) is common.
 Laboratory findings
Laboratory findings
The white blood cell count is typically elevated with a left shift. In the absence of prior renal insufficiency, renal function is initially normal, but blood urea nitrogen and creatinine rise progressively. Hepatic transaminases may be markedly elevated due to liver hypoperfusion. Poor tissue perfusion may result in an anion-gap acidosis and elevation of the lactic acid level. Before support with supplemental O2, arterial blood gases usually demonstrate hypoxemia and metabolic acidosis, which may be compensated by respiratory alkalosis. Cardiac markers, creatine phosphokinase and its MB fraction, and troponins I and T are markedly elevated.
 Electrocardiogram
Electrocardiogram
In CS due to acute MI with LV failure, Q waves and/or >2-mm ST elevation in multiple leads or left bundle branch block are usually present. More than one-half of all infarcts associated with shock are anterior. Global ischemia due to severe left main stenosis usually is accompanied by severe (e.g., >3 mm) ST depressions in multiple leads.
 Chest roentgenogram
Chest roentgenogram
The chest x-ray typically shows pulmonary vascular congestion and often pulmonary edema, but these findings may be absent in up to a third of patients. The heart size is usually normal when CS results from a first MI but is enlarged when it occurs in a patient with a previous MI.
 Echocardiogram
Echocardiogram
A two-dimensional echocardiogram with color-flow Doppler (Chap. 12) should be obtained promptly in patients with suspected CS to help define its etiology. Doppler mapping demonstrates a left-to-right shunt in patients with VSR and the severity of MR when the latter is present. Proximal aortic dissection with aortic regurgitation or tamponade may be visualized, or evidence for pulmonary embolism may be obtained.
 Pulmonary artery catheterization
Pulmonary artery catheterization
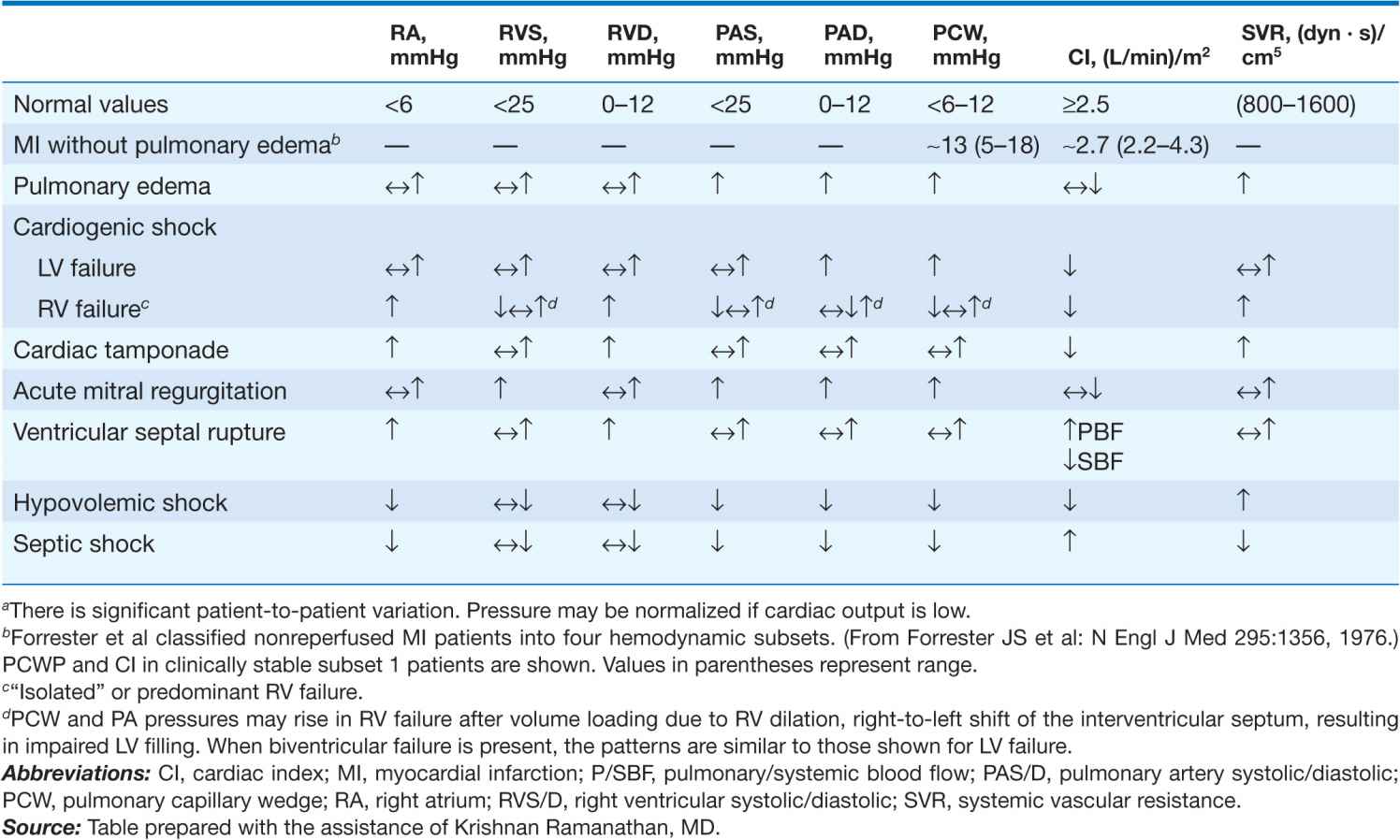
There is controversy regarding the use of pulmonary artery (Swan-Ganz) catheters in patients with established or suspected CS (Chap. 13). Their use is generally recommended for measurement of filling pressures and cardiac output to confirm the diagnosis and optimize the use of IV fluids, inotropic agents, and vasopressors in persistent shock (Table 28-2). Blood samples for O2 saturation measurement should be obtained from the right atrium, right ventricle, and pulmonary artery to rule out a left-to-right shunt. Mixed venous O2 saturations are low and arteriovenous (AV) O2 differences are elevated, reflecting low cardiac index and high fractional O2 extraction. However, when a systemic inflammatory response syndrome accompanies CS, AV O2 differences may not be elevated. The PCWP is elevated. However, use of sympathomimetic amines may return these measurements and the systemic BP to normal. Systemic vascular resistance may be low, normal, or elevated in CS. Equalization of right- and left-sided filling pressures (right atrial and PCWP) suggests cardiac tamponade as the cause of CS (Chap. 22).
TABLE 28-2
HEMODYNAMIC PATTERNSa
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


