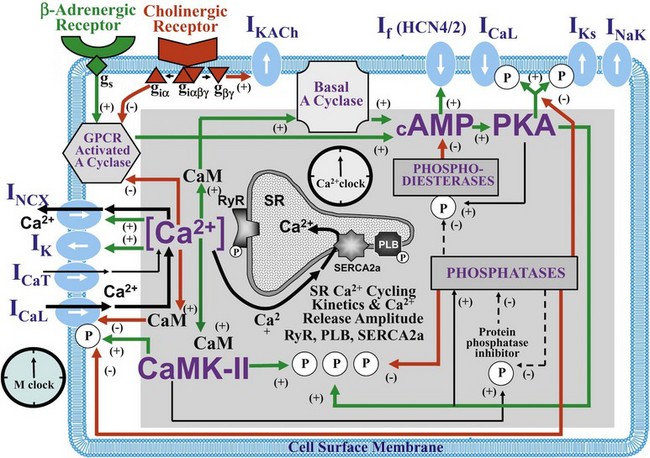
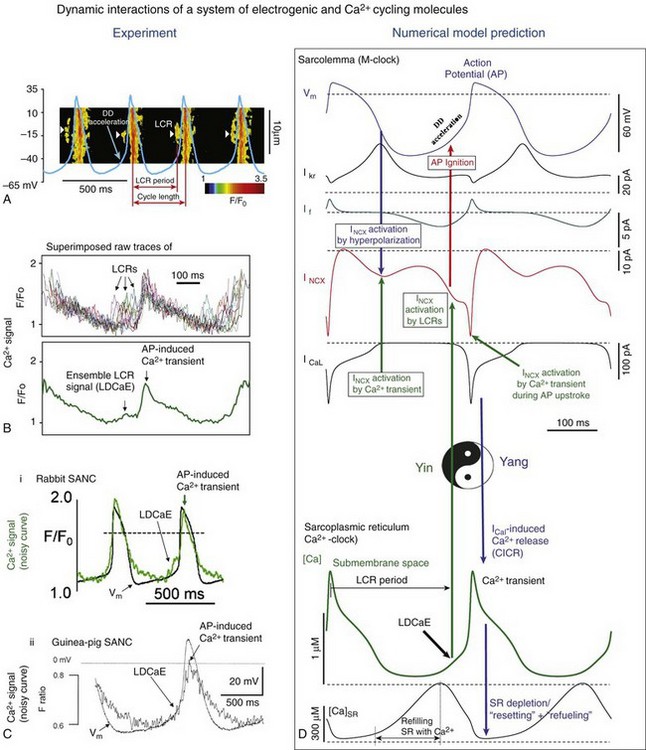
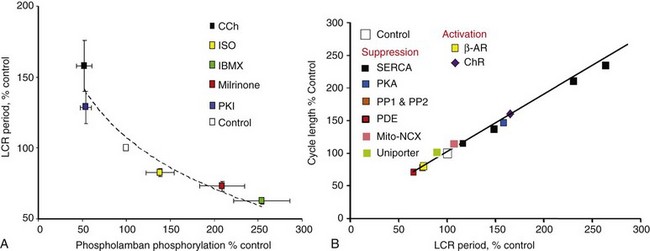
25 Realization of Importance of Ca2+ Signaling for Cardiac Pacemaker Function Ca2+-Clocks in Pacemaker Cells Crosstalk of Ryr and NCX to Transfer Intracellular Ca2+ Signals to M-Clock The Essence of Pacemaker Function Is a Coupled Function of an Intracellular Ca2+-Clock and Membrane Ion Channel Clock Autonomic Receptor Modulation of the Coupled-Clock System Mitochondrial Function in SANC An Additional Level of Complexity of Heart Pacemaker Function Arises Within the SAN Tissue The present perspective views SANC, per se, as a system comprising several levels of complexity and integrated components. Sarcolemmal ion currents (Figure 25-1 and Table 25-1) have been extensively studied in the past (see review1), and the ensemble of these currents re-created in silico from experimental voltage-clamp data can generate spontaneous APs.2 The subsystem of sarcolemmal molecules forming a voltage membrane oscillator was conceptualized as a membrane clock3 (M-clock, for short) and has been the predominant feature in at least 12 SANC numerical models.2 During rhythmic spontaneous AP firing by SANC, the M-clock is dynamically coupled to Ca2+ cycling (see Figure 25-1, gray area) via multiple voltage-, Ca2+-, cyclic adenosine monophosphate (cAMP)-, and phosphorylation-dependent mechanisms (coupling factors).4 SANC sarcoplasmic reticulum (SR), however, in isolation from the M-clock can generate its own spontaneous local (submembrane) rhythmic Ca2+ signals. Thus, similar to the M-clock, the SR is intrinsically also an almost perfectly periodic oscillator that can be envisioned as a Ca2+-clock.3 During spontaneous AP firing by SANC, the M- and Ca2+-clocks do not exist as separate entities: They mutually entrain each other via aforementioned coupling factors. These coupled oscillators that differ in nature thus represent heterogeneous redundancy within SANC that confers a physiological robust, yet extremely flexible, united pacemaker cell system.4,5 Figure 25-1 Schematic illustration of interactions of identified molecules comprising the fully coupled-pacemaker cell clock. Note that common regulatory factors (purple large lettering) govern the function of both the SR Ca2+-clock (gray intracellular area) and the M-clock (light-blue cell membrane area with blue labels depicting electrogenic proteins). These common factors act as nodes within the system to couple the function of the activities of both clocks. The system is balanced as illustrated by traffic light–like colors: Green arrows designate signaling driving AP firing, but red arrows show suppression, maintaining a given steady state level of cAMP and protein phosphorylation. G protein–coupled receptors (green and red shapes within the membrane) modulate both Ca2+-clock and M-clock function via those same crucial signaling nodes of the system. (Modified from Lakatta EG, Maltsev VA, Vinogradova TM: A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 106:659–673, 2010.) In the late 1970s to the early 1980s, as the importance of an intracellular Ca2+ transient (as the trigger for ventricular myocyte contraction) came into focus, the idea that an intracellular Ca2+ oscillator could also drive membrane excitations in Purkinje fibers was suggested.6 But because oscillatory current in Purkinje fibers became manifest only in a Ca2+ overload state, such current fluctuations were interpreted in the context of “abnormal automaticity” (i.e., they were attributed to a disturbance in normal cardiac function7). In the late 1980s, afterdepolarizations and aftercontractions were recorded in Purkinje fibers under normal Ca2+ loading conditions,8 and these fibers demonstrated localized, spontaneous myofilament motion caused by spontaneous local Ca2+ oscillations9 in the absence of Ca2+ overload. A cytosolic Ca2+ transient is evoked by each spontaneous SANC AP (see Figure 25-2, D), and that Ca2+ influx via L-type Ca2+ channels (LCCh) (see Figure 25-1) affects SR Ca2+ loading. Chelation of intracellular Ca2+ in SANC10–12 markedly slowed or abolished spontaneous AP firing. The importance of SR Ca2+ release and Na+/Ca2+ exchanger (NCX) current (INCX) for pacemaker cell pacemaker function was also demonstrated directly by effects of ryanodine on Ca2+ cycling.13,14 Ryanodine (by disabling the ryanodine receptor [RyR] and the SR release channel [Figure 25-2], and by depleting SR Ca2+ content] had a profound negative chronotropic effect on the automaticity of subsidiary atrial pacemakers15 and in SANC.13,14 Additional voltage-clamp experiments in perforated patch configuration in isolated cat atrial latent pacemaker cells demonstrated the occurrence of an inward current during late DD that was sensitive to ryanodine, and presumably was linked to INCX activated by SR Ca2+ release.16 Figure 25-2 A, Line-scan image of LCRs (white arrowheads) with superimposed spontaneous APs recorded in rabbit SANC. B, Upper panel, LCRs imaged by confocal microscopy; lower panel, Temporal average of LCRs creates ensemble LCR signal or late diastolic Ca2+ elevation (LDCaE) that precedes AP-induced Ca2+ transient.29 C, LDCaE in single SANC of rabbit57 and guinea pig.20 D, Novel numerical model predicts “yin-yang” types of interactions within the coupled-clock system of SANC (see text for details). D, Diastolic depolarization; SR, sarcoplasmic reticulum. (Modified from Maltsev VA, Lakatta EG: Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am J Physiol Heart Circ Physiol 296:H594–H615, 2009.) Confocal imaging of Ca2+ in mammalian SANC and atrial subsidiary pacemaker cells combined with noninvasive perforated patch-clamp electrophysiology17,18 and imaging of toad sinus venosus cells19 has documented the occurrence of spontaneous, localized, diastolic Ca2+ release in pacemaker cells in the absence of Ca2+ overload. These local Ca2+ releases (LCRs) during DD are initiated beneath the cell surface membrane via spontaneous activation of RyR (Figure 25-2A,B). SANC exhibit robust SERCA2 and RyR immunolabeling,20,21 and although SERCA2 immunolabeling in SANC occurs diffusely throughout the cytoplasm and in perinuclear area, RyR immunolabeling is most intense in the subsarcolemmal space.20,21 LCRs appear as 4- to 10-µm Ca2+ wavelets in confocal line-scan images of spontaneously firing rabbit SANC and emerge following dissipation of the global systolic transient effected by the previous AP; they crescendo during DD, peaking during late DD, as they merge into the global cytosolic Ca2+ transient triggered by the next AP (see Figure 25-2A).18,22 LCRs and the ensemble LCR signal (i.e., integral of all LCRs), that is, late diastolic Ca2+ elevations (LDCaE), have now been documented in numerous species (see review4) (see Figure 25-2, B-D). LCR occurrence does not require triggering by depolarization of the surface membrane: Persistent rhythmic oscillatory membrane currents can be activated by rhythmic LCRs during voltage-clamp (at potentials that prevent cell Ca2+ depletion, e.g., −10 mV). Both persistent LCRs and the currents activate simultaneous fluctuations of the same frequency,22,23 and both are abolished by ryanodine. Sustained LCR activity is also observed in chemically “skinned” SANC (i.e., having a detergent-permeabilized cell surface membrane) bathed in a physiological [Ca2+] of 100 nM.22,23 Because LCRs are generated as rhythmic events at rates of 1 to 5 Hz (i.e., encompassing those of spontaneous AP firing in SANC), SR was conceptualized as an intracellular “Ca2+-clock.”3 The ability of SANC SR to generate sustained intracellular Ca2+ oscillations under normal physiological conditions within this broad frequency range has also been demonstrated in numerical model simulations that have embraced the occurrence of experimentally determined LCRs.5 RyR2 knockout not only causes failure of spontaneous Ca2+ releases in embryonic cardiac cells and suppresses spontaneous cell beating, it also leads to a marked depression of the obligatory developmental increase in heart rate and cardiac output that, in the absence of knockout, support continued cardiac embryonic differentiation.24 As in adult SANC, a crucial regulatory role of RyR2-mediated Ca2+ releases in pacemaker function is demonstrated in RyR2 knockout cells, as β-adrenergic receptor (β-AR) stimulation is ineffective in these cells.25 An essential role of RyR2 in pacemaker function has been recently demonstrated in tissue-specific RyR2 knockout mice with acute ≈50% loss of RyR2 protein in the heart, but not in other tissues.26 The RYR2 loss-of-function causes bradycardia and arrhythmia. Furthermore, cardiac RyR2 knockout mice exhibit some functional and structural hallmarks of heart failure, including sudden cardiac death. NCX is not an ion channel, and its amplitude almost instantly follows changes in membrane potential or intracellular [Ca2+]. NCX function is both voltage- and Ca2+-dependent. When intracellular [Ca2+] is high as the result of AP-induced Ca2+ transient, AP repolarization activates NCX forward mode, generating inward current INCX by exchanging one Ca2+ (going out) to 3 Na+ (going into the cell). In contrast to hyperpolarization-activated “funny” current, If (see Table 25-1), INCX is activated earlier, as it almost instantly follows membrane hyperpolarization (see Figure 25-2, D). Furthermore while the non-selective If is decreasing and reversing during later DD, INCX is substantially increasing further by waxing diastolic Ca2+ release from the SR (see Figure 25-1 and Figure 25-2, D, described later in detail). Thus, INCX has been realized as a major transduction mechanism of intracellular Ca2+ signaling that crucially contributes to DD and normal automaticity of SANC. The NCX is one of the earliest functional genes that exist during early embryonic heart development,27 and NCX1 is required for normal pacemaker activity. Atrial-specific NCX knockout mice that lack NCX1 but have intact If, and express NCX1 in ventricular myocytes,28 live to adulthood, and exhibit a markedly reduced heart rate. However APs can be evoked under current-clamp conditions in SANC isolated from these mice, indicating that knockout cells retain electrical excitability. At the resolution of the confocal microscope21 NCX and RyRs, molecules appear co-localized across the ≈12-nm subsarcolemmal gap between RyRs on the SR and NCX molecules on the sarcolemma. The crosstalk of LCRs and NCX activates a forward mode NCX operation that generates local inward INCX currents, which produce miniature membrane voltage fluctuations.18,29,30 The net late DD inward current initiated by the LCR ensemble (measured as ryanodine-sensitive current) in rabbit SANC varies from 0.3 pA/pF31 to 1.6 pA/pF,18 yielding a whole-cell INCX range from 9.6 to 51.2 pA for a 32-pF SANC (see Figure 25-2, D). Because an extremely small net ion current change (a few pA in rabbit SANC) has marked effects on membrane potential, an inward current of this magnitude is sufficient to broadly modulate the DD.5,30,31 For comparison, the peak of If achieved during DD in SANC ranges from 0.02 to 0.23 pA/pF as predicted by 12 different numerical models.2 LCR-activated inward INCX, together with other DD mechanisms (such as L-type Ca2+ current, ICaL), imparts the exponentially rising phase to the DD18,29,30 that initiates the subsequent AP (see Figure 25-2, A-C). Although voltage-activated L-type Ca2+ currents surely contribute to late DD, and L-type Ca2+ currents are required for AP initiation, failure to generate diastolic INCX signals results in pacemaker failure when ICaL density is normal (e.g., when RyRs are disabled ryanodine or during short-term NCX blockade18). Thus available experimental and numerical modeling data clearly indicate an important role of Ca2+ cycling molecules and their “crosstalk” in normal physiological function. The LCR period is the delay between the AP-triggered global cytosolic Ca2+ transient and LCR emergence during DD (Figure 25-2, A). It reports the time at which AP ignition is prompted via INCX activation, and it closely predicts the AP cycle length in numerous experimental perturbations of SANC function (Figure 25-3, B). Over the wide range of conditions and widely varying AP firing rates from about 1 to 4 Hz, the relationship of LCR periods to the spontaneous cycle lengths of SANC is nearly identical, with a slope being close to 1 and an offset “ignition” time of 10 to 100 ms (review4). The LCR period and the cycle length remain strongly coupled, not only in the steady state beating, but also during stringent transitions, such as the transient state after removal of voltage-clamp at the maximum diastolic potential (MDP),22 and during intrinsic beat-to-beat variations in AP cycle length.32 Figure 25-3 A, PKA-dependent changes in phosphorylation of phospholamban predict changes in the SANC LCR period. B, Changes in the steady state LCR period in response to numerous perturbations of the coupled-clock system predict concomitant changes in the steady state spontaneous AP cycle length. (Modified from Lakatta EG, Maltsev VA, Vinogradova TM: A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 106:659-673, 2010. Updated with data from Vinogradova TM, Brochet DX, Sirenko S, et al: Sarcoplasmic reticulum Ca2+ pumping kinetics regulates timing of local Ca2+ releases and spontaneous beating rate of rabbit sinoatrial node pacemaker cells. Circ Res 107:767–775, 2010.)
Cardiac Impulse Is Initiated by a Coupled System of Membrane Ion Channels and Ca2+ Cycling Proteins
Realization of Importance of Ca2+ Signaling for Cardiac Pacemaker Function
Ca2+-Clocks in Pacemaker Cells
Crosstalk of Ryanodine Receptor and Na+/ Ca2+ Exchanger to Transfer Intracellular Ca2+ Signals to M-Clock
The Essence of Pacemaker Function Is a Coupled Function of an Intracellular Ca2+-Clock and Membrane Ion Channel Clock
The LCR Period Reports the Functional State of the Coupled System (Not That of the Ca2+-Clock Per Se)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cardiac Impulse Is Initiated by a Coupled System of Membrane Ion Channels and Ca2+ Cycling Proteins

