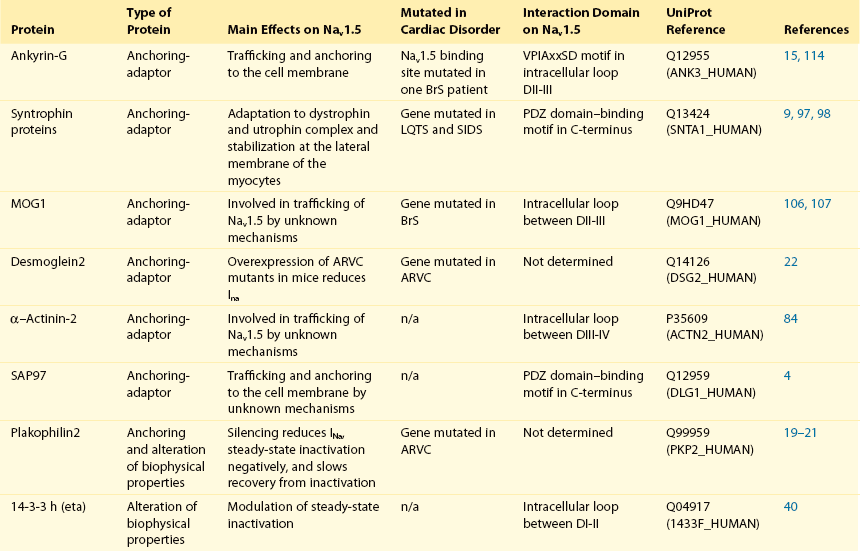
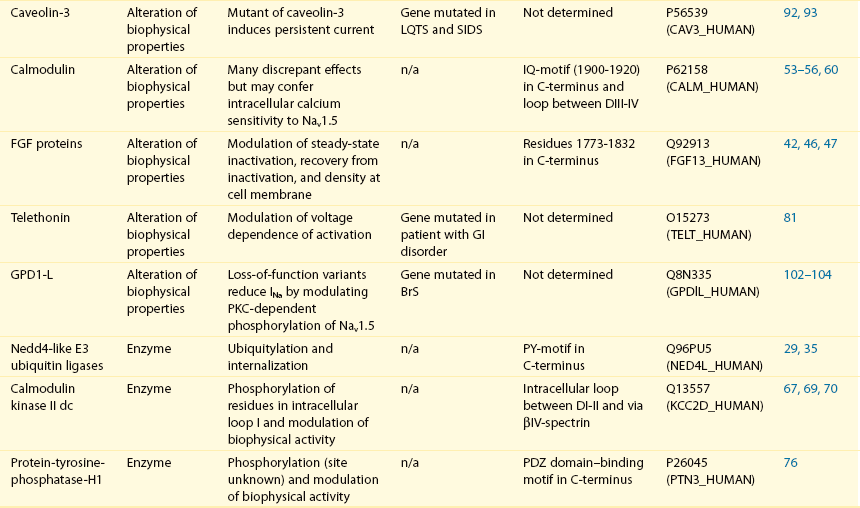
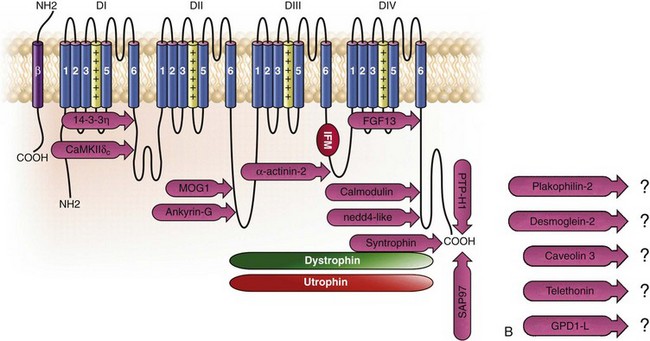
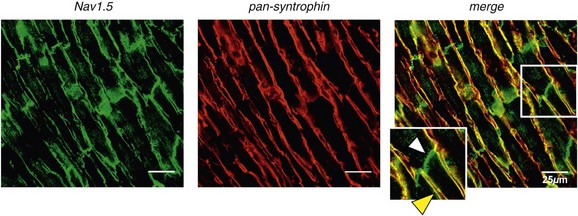
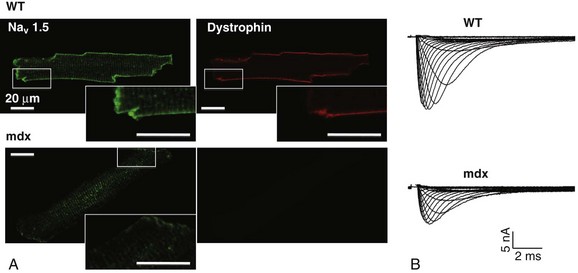
18 Among the many ionic currents known to be involved in the genesis of the cardiac action potential (AP), the Na+ current (INa) has been studied for more than 50 years1 and remains a major focus of research. The basic structure and function of the cardiac Na+ channel Nav1.5 and its central role in cardiac pathologies are covered in Chapters 1, 9, and 50 of this book. This chapter summarizes recently published data on the proteins interacting with Nav1.5 that form distinct macromolecular and multiprotein complexes. The roles of the four β-subunits are not reviewed because that topic is covered in other chapters of this book. Over the past several years, Nav1.5 has been shown to interact with a growing list of regulatory proteins (Figure 18-1, A and B; Table 18-1). The genes coding for several of these interacting proteins have been found in patients with inherited arrhythmias, such as congenital long QT syndrome (LQTS)2 and Brugada syndrome (BrS).3 The proteins interacting with Nav1.5 have been classified as (1) anchoring/adaptor proteins involved in trafficking, targeting, and anchoring the channel protein to specific membrane compartments; (2) enzymes interacting with and modifying the channel structure via posttranslational modifications such as protein kinases or ubiquitin ligases; and (3) proteins modulating the biophysical properties of Nav1.5 upon binding (see Table 18-1). These classifications are not mutually exclusive. Table 18-1 The 16 Proteins (or Families of Proteins) Reported to Interact and Regulate Nav1.5 This table does not take into account the four β sodium channel subunits. Figure 18-1 Schematic representation of the α- and β-subunits of the Nav1.5 and interacting proteins. A, The predicted membrane topology of the α-subunit of Nav1.5 is illustrated. DI-DIV indicates the four homologous domains of the α-subunit; segments S5 and S6 are the pore-lining segments and the S4 helices (yellow) serve as voltage sensors. In the connecting loop between DIII and DIV, the IFM red symbol presents the three residues (isoleucine, phenylalanine, and methionine) known to play a key role in the fast inactivation process. The two intracellular loops display many sites for phosphorylation, whereas the C-terminal domain bears many protein-protein interaction motifs. Only one β-subunit of the four known to be able to interact with sodium channels is shown (purple = transmembrane domain protein on the left side).The eleven proteins, in addition to the β-subunits, are reported to interact with Nav1.5, for which a binding site to one of the intracellular domains is represented schematically. B, For five proteins, the interaction has been shown only by coimmunoprecipitation experiments, but the binding sites are still unknown (question marks). These interactions may be indirect (i.e., requiring adaptor proteins). Immunofluorescence staining experiments have shown that Nav1.5 is expressed in distinct membrane compartments (i.e., the intercalated discs and the “lateral membrane” of cardiac cells) (Figure 18-2).4–6 This notion has been under debate because earlier studies described a quasi-exclusive localization of Nav1.5 at the intercalated discs.7,8 Our group proposed a model of Nav1.5 localization at two distinct pools based on strong molecular and in vivo evidence showing that Nav1.5 belongs to the dystrophin multiprotein complex.9 Dystrophin and syntrophin proteins are not expressed at the intercalated discs of human,10 rat,11 or mouse cardiomyocytes.4 Recent studies have presented evidence supporting this multiple-pool model of Nav1.5. Roden’s group investigated a knock-in mouse model harboring the p.D1275N mutation in the gene SCN5A, which codes for Nav1.5 and is found in patients with dilated cardiomyopathy.12 They observed a marked reduction in the expression of Nav1.5 exclusively at the lateral membrane.12 More recently, Lin et al.13 obtained functional evidence for two pools of cardiac Na+ channels with different biophysical properties by performing macropatch experiments at different locations in cardiac cells. It is very possible that this two-pool expression model of Nav1.5 is an oversimplification because there is both functional14 and morphologic evidence6,15 of a T-tubular population of Nav1.5 (Figure 18-3). In a recent paper, Nav1.5 was found to be colocalized with SAP97 at the T-tubules.16 Our recent findings suggest that this third pool does not depend on the syntrophin/dystrophin complex because it is well distinguished in dystrophin-deficient (mdx) myocytes (see Figure 18-3). Figure 18-2 Stainings of cardiac sections leading to the concept of multiple pools of Nav1.5 channels in cardiac cells. Sections of mouse ventricular myocardium with anti-Nav1.5 staining (left panel), anti-pan-syntrophin staining (middle panel), and overlay (right panel, merge). From these stainings, it is clear that syntrophin proteins are not expressed at the intercalated discs (white arrowhead) where Nav1.5 is present. This defines the intercalated disc pool of Nav1.5 that has been proposed to interact with SAP97 (see Figure 1, A, and Figure 4). The yellow arrowhead shows the lateral membrane pool of Nav1.5 colocalized with syntrophin proteins; white bar = 25 µm. Figure 18-3 Reduction of Nav1.5 and sodium current in dystrophin-deficient mouse cardiomyocytes. A, Isolated mouse cardiomyocytes from wild type and mdx (dystrophin-deficient) mice with Nav1.5 staining (green) and dystrophin staining (red). Whereas Nav1.5 channels are found at the intercalated discs and at the lateral membrane compartment, it is apparent that dystrophin is excluded from the intercalated discs. The Nav1.5 pool at the discs has been shown to colocalize with SAP97. B, Sodium current recordings from whole-cell patch-clamp experiments of freshly isolated wild type and mdx mouse cardiomyocytes. It can be inferred that the reduction in current is caused by the specific loss of Nav1.5 channels from the lateral membrane pool. (A, From Petitprez S, Zmoos AF, Ogrodnik J, et al: SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108:294–304, 2011; with permission from Wolters Kluwer Health. B, Modified from Gavillet B, Rougier JS, Domenighetti AA, et al: Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res 18:407–414, 2006; with permission from Wolters Kluwer Health.) The relationship between the expression of Nav1.5 at the intercalated discs and other disc proteins has recently been studied. It was observed that the intercalated disc pool of Nav1.5 is dependent on the expression of key proteins known to be well expressed at the discs, such as connexin43,17,18 plakophilin2,19–21 and desmoglein2.22 Thus, Nav1.5 not only interacts with many different partners, but its localization in different cellular and membrane compartments is also very diverse. One of the obvious conclusions of these observations is that there is not one cardiac Na+ channel Nav1.5, but rather a multiplicity of them with variable functions and regulatory mechanisms. This section lists the proteins that interact with Nav1.5 that currently have no demonstrable pathologic roles. These interacting proteins were discovered by either performing protein-protein interaction screens, such as yeast two-hybrid assays, or by using proteomic-based protein identification assays. The sites of interaction, often protein-protein interaction domains, were mapped on the sequence of Nav1.5 as described in Figure 18-1, A. This list does not follow any specific logic related to importance but is dictated by the chronologic order of the published studies. Ubiquitylation of target proteins tags them for proteasome- or lysosome-dependent degradation23 but also serves multiple nondegradative functions, in particular the trafficking of membrane proteins.24 Ubiquitin is a small protein of 76 amino acids found in all animal cells.25 It binds covalently to lysine residues of target proteins. This protein ubiquitylation is performed by E3 ubiquitin-protein ligases.25 Ubiquitylated membrane proteins are subsequently internalized and can be targeted for lysosomal or proteasomal degradation. Alternatively, they can also be deubiquitylated by specific proteases and recycled back to the membrane.26,27 Many different ion channels have been reported recently to be regulated by the Nedd4-like family of E3 ubiquitin-protein ligases. Nedd4-like enzymes bind specifically to target proteins that have consensus domains known as PY motifs with the sequence [L/P]PxY.28 Such PY motifs are found in the C-termini of all voltage-gated Na+ channels (with the exception of Nav1.4, Nav1.9, and Nax29,30), as well as in other cardiac ion channels.27,31,32 Nedd4/Nedd4-like enzymes harbor several WW domains33 that can interact with these PY motifs. When expressed in Xenopus oocytes, Nav1.5-mediated INa is decreased by Nedd4-2–mediated ubiquitylation.34 Our group investigated the molecular determinants of this regulation35 and found that the ubiquitin-protein ligase Nedd4-2 binds directly to the PY motif of Nav1.5 and ubiquitylates the channel in mammalian cells. Because there was no reduction in the total level of Nav1.5 protein upon coexpression with Nedd4-2, we concluded that this was most likely caused by an increased internalization rate rather than by Nav1.5 degradation.29 Inhibition of the proteasome has been linked recently to an increase in INa and Nav1.5 expression in neonatal rat cardiomyocytes.36 Ubiquitylated Nav1.5 was found to be present in mouse cardiac tissue,35 further suggesting that membrane turnover or the stability of Nav channels can be regulated in vivo via ubiquitylation. Nine of such Nedd4-like E3 enzymes are present in the human genome,37 at least eight of which are expressed at the RNA level in human cardiac tissue.31 It remains to be determined which of these Nedd4/Nedd4-like proteins regulates Nav1.5 in a physiologic context and whether the PY motif plays a role in cardiac disease. Altogether these results suggest that the ubiquitin-proteasome system is involved in several aspects of Nav1.5 regulation, the intricacies of which remain to be elucidated. The 14-3-3 protein family is composed of dimeric cytosolic adaptor ubiquitous proteins.38 The members of this family are involved in many cellular functions such as the binding and regulation of trafficking of various membrane proteins.39 Allouis et al. performed yeast two-hybrid and coimmunoprecipitation experiments40 showing that the isoform 14-3-3η interacts with the N-terminal part of the intracellular loop linking domains I to II (see Figure 18-1, A and Table 18-1). Furthermore, it was observed that 14-3-3 and Nav1.5 are colocalized at the intercalated discs of myocytes. No influence on the Nav1.5-mediated peak current was observed when Nav1.5 and 14-3-3η were coexpressed in COS cells, suggesting that this protein does not influence Nav1.5 trafficking. In this expression system, 14-3-3 shifted the inactivation curve toward negative potentials and delayed recovery from inactivation, illustrating that 14-3-3 proteins are able to modify the biophysical properties of ion channels. Because different isoforms of 14-3-3 proteins are expressed in cardiac cells,40 their exact roles in normal cardiac function and their implications in disease states require further investigation. Although fibroblast growth factor homologous factor (FHF) family members are highly homologous in sequence and structure to fibroblast growth factors (FGF), their functional roles are quite different. FHFs are found in the cytosol because their N-termini lack signal sequences that are required for secretion.41 The FGF homologous factors FHF3, FHF1, FHF2, and FHF4 are also called FGF11, FGF12, FGF13, and FGF14, respectively. One of the best characterized functions of the FHFs is their ability to bind to and modulate voltage-gated Na+ channels. Liu et al.42 were the first to demonstrate that FGF12 interacts with the proximal portion of the C-terminus of Nav1.5 (see Figure 18-1, A). This finding was then confirmed by Goetz et al.,43 who also showed that FGF11 and FGF14 interact with the same domain of Nav1.5. In HEK293 cells, coexpression of FGF12 with Nav1.5 shifted the steady-state inactivation relationship toward hyperpolarized values without affecting the other parameters studied.42 Interestingly, several LQTS type 3 and BrS mutations are located in the domain that interacts with FHF1B (see Table 18-1). The SCN5A mutation D1790G44 disrupted the binding of this protein with Nav1.5 and abolished the FHF 12-induced shift of steady-state inactivation. Another member of the FGF family, FGF14, has been shown to downregulate the activity of Nav1.5.45 Because this regulatory protein is expressed in the brain, and not in the heart,45 the significance of this interaction is likely restricted to the central nervous system. A recent study46 demonstrated that FGF13 is expressed in mouse cardiac myocytes. FGF13 was shown to directly bind to and colocalize with Nav1.5 in mouse cardiac cells. Reducing the expression of FGF13 decreased the Na+ current, shifted the availability curve toward more negative potentials, and slowed recovery from inactivation. Furthermore, decreased membrane expression of Nav1.5 was observed in cells with reduced expression of FGF13. These results underscore the specific role of FGF13 in modulating Nav1.5 function and suggest that this gene may be a susceptibility gene for cardiac arrhythmias. A crystal structure of the Nav1.5 C-terminal domain complexed with FGF13 and calmodulin was recently published.47 The functional relationship between FGFs and calmodulin remains to be studied in more detail. Intracellular Ca2+ has been shown to modulate the function of many ion channels, including the voltage-gated Na+ channels.48,49 Many cardiac ion channels use calmodulin (CaM), a ubiquitous intracellular Ca2+-binding protein involved in many different cellular processes,50 as a Ca2+-sensing partner. The Nav1.5 C-terminal domain has an IQ motif with a consensus sequence of IQxxxRxxxxR (see Table 18-1), which is very similar to that found in voltage-gated calcium channels.51 The IQ motif is also found in all isoforms of the Nav family.52 Several studies47,53–55 have shown a direct interaction between CaM and the IQ motif of Nav1.5. Recent work by Wang et al. described a crystal structure with an IQ motif ternary complex of Nav1.5, FGF13, and CaM. The functional consequences of this CaM-Nav1.5 interaction are controversial. Several studies52,56,57 have shown inconsistent results that have been difficult to reconcile. A few groups53,55–57 reported that the voltage dependence and stability of the inactivated state were dependent on CaM and the IQ motif. In addition, it has been proposed that CaM may not be the only sensor for the Ca2+-dependent regulation of Nav1.5, because the proximal part of the Nav1.5 C-terminal domain is similar to an EF-hand motif, which is known to bind Ca2+.58,59 Whether this domain binds intracellular calcium is still under debate.47,60–62 The role of intracellular Ca2+ in Nav1.5 regulation was recently investigated by Casini et al.63 They demonstrated that Nav1.5 single-channel conductance is decreased (most likely directly) with an increase in the intracellular Ca2+ concentration. Intracellular Ca2+ appears to be an important regulator of Nav1.5 function, but more studies are needed to clarify its role. Ca2+/calmodulin-dependent protein kinases II (CaMKII) are serine/threonine protein kinases expressed in many cell types, where they transduce intracellular Ca2+ increases into the phosphorylation of target proteins, including cardiac ion channels.64 CaMKIIδc is the predominant cardiac isoform upregulated in human and animal heart failure models.65,66 Nav1.5 has been found to colocalize and coimmunoprecipitate with CaMKIIδc.67–69 Ashpole et al.69 have shown that CaMKIIδc interacts with the first intracellular loop of Nav1.5 (see Figure 18-1) and that the residues Ser-516 and Thr-594 may be phosphorylated by this kinase. Hund et al.70 found that Ser-571 was also a target of CaMKII within the same intracellular loop of Nav1.5, and that this regulation was dependent on the correct expression of βIV-spectrin, which interacts with ankyrin-G at the intercalated discs. Overexpression of the CaMKIIδc enzyme in rabbit myocytes and transgenic mice induced a Ca2+-dependent hyperpolarizing shift of the steady-state inactivation curve, slowed the recovery from inactivation, and increased the persistent INa. These biophysical property alterations of INa are similar to some congenital LQTS type 3 mutation-dependent modifications.71 In mice, transgenic overexpression of CaMKIIδc leads to chronic heart failure and episodes of ventricular tachycardia. In dog ventricular myocytes, it has also been shown that CaMKII activity increases the persistent/late INa,72 but it is unclear whether these arrhythmias are the direct consequence of the Nav1.5 biophysical alterations or are related to other heart failure mechanisms. Nevertheless, CaMKII is an important component in the pathogenesis of arrhythmias and potentially a new drug target.64 Ion channels are also regulated by the phosphorylation of tyrosine residues, a process that depends on both the phosphorylation activity of tyrosine protein kinases and the dephosphorylation activity of phosphatases.73 It was demonstrated that the overexpression of the protein tyrosine kinase Fyn in HEK293 cells altered several of the biophysical properties of Nav1.5 via the phosphorylation of Tyr-1495.74
Macromolecular Complexes and Regulation of the Sodium Channel Nav1.5
Nav1.5 and Interacting Proteins
Localization of Nav1.5 in Cardiac Cells: Evidence for Distinct Pools
Proteins Interacting with Nav1.5 Without Demonstrated Roles in Arrhythmias
Ubiquitin-Protein Ligases of the Nedd4/Nedd4-like Family
14-3-3η (eta) Protein
Fibroblast Growth Factor Homologous Factors
Calmodulin
Ca2+/Calmodulin-dependent Protein Kinase II
Proteins Tyrosine Phosphatase PTPH1
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Macromolecular Complexes and Regulation of the Sodium Channel Nav1.5
