BASIC BIOLOGY OF THE CARDIOVASCULAR SYSTEM
Joseph Loscalzo ![]() Peter Libby
Peter Libby ![]() Jonathan Epstein
Jonathan Epstein
THE BLOOD VESSEL
VASCULAR ULTRASTRUCTURE
Blood vessels participate in homeostasis on a moment-to-moment basis and contribute to the pathophysiology of diseases of virtually every organ system. Hence, an understanding of the fundamentals of vascular biology furnishes a foundation for understanding the normal function of all organ systems and many diseases. The smallest blood vessels—capillaries—consist of a monolayer of endothelial cells apposed to a basement membrane, adjacent to occasional smooth-muscle-like cells known as pericytes (Fig. 1-1A). Unlike larger vessels, pericytes do not invest the entire microvessel to form a continuous sheath. Veins and arteries typically have a trilaminar structure (Fig. 1-1 B–E). The intima consists of a monolayer of endothelial cells continuous with those of the capillaries. The middle layer, or tunica media, consists of layers of smooth-muscle cells; in veins, the media can contain just a few layers of smooth-muscle cells (Fig. 1-1 B). The outer layer, the adventitia, consists of looser extracellular matrix with occasional fibroblasts, mast cells, and nerve terminals. Larger arteries have their own vasculature, the vasa vasorum, which nourishes the outer aspects of the tunica media. The adventitia of many veins surpasses the intima in thickness.
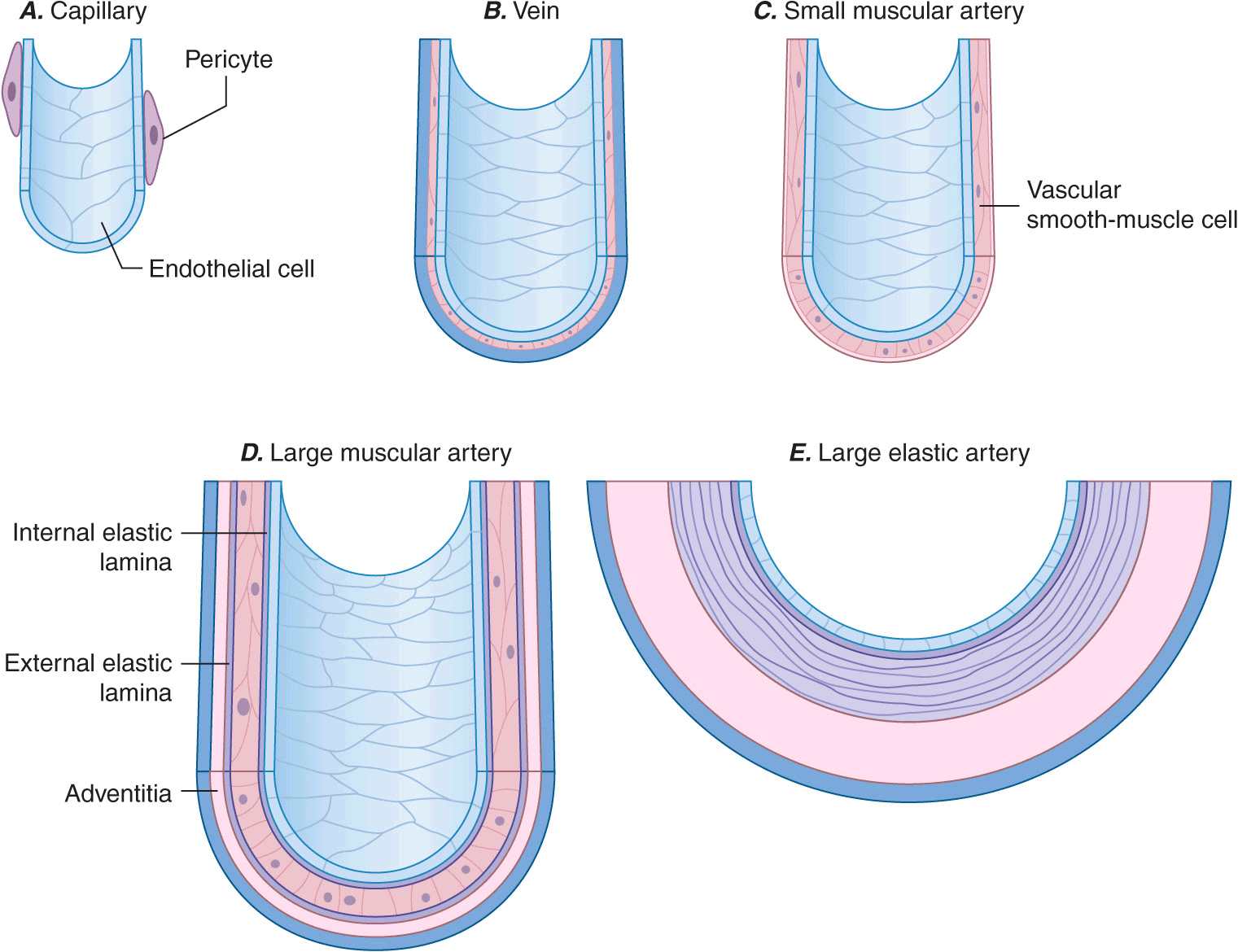
FIGURE 1-1
Schematics of the structures of various types of blood vessels. A. Capillaries consist of an endothelial tube in contact with a discontinuous population of pericytes. B. Veins typically have thin medias and thicker adventitias. C. A small muscular artery features a prominent tunica media. D. Larger muscular arteries have a prominent media with smooth-muscle cells embedded in a complex extracellular matrix. E. Larger elastic arteries have cylindrical layers of elastic tissue alternating with concentric rings of smooth-muscle cells.
The tone of muscular arterioles regulates blood pressure and flow through various arterial beds. These smaller arteries have a relatively thick tunica media in relation to the adventitia (Fig. 1-1 C). Medium-size muscular arteries similarly contain a prominent tunica media (Fig. 1-1 D); atherosclerosis commonly affects this type of muscular artery. The larger elastic arteries have a much more structured tunica media consisting of concentric bands of smooth-muscle cells, interspersed with strata of elastin-rich extracellular matrix sandwiched between layers of smooth-muscle cells (Fig. 1-1 E). Larger arteries have a clearly demarcated internal elastic lamina that forms the barrier between the intima and the media. An external elastic lamina demarcates the media of arteries from the surrounding adventitia.
ORIGIN OF VASCULAR CELLS
The intima in human arteries often contains occasional resident smooth-muscle cells beneath the monolayer of vascular endothelial cells. The embryonic origin of smooth-muscle cells in various types of arteries differs. Some upper-body arterial smooth-muscle cells derive from the neural crest, whereas lower-body arteries generally recruit smooth-muscle cells from neighboring mesodermal structures during development. Derivatives of the proepicardial organ, which gives rise to the epicardial layer of the heart, contribute to the vascular smooth-muscle cells of the coronary arteries. Recent evidence suggests that bone marrow may give rise to both vascular endothelial cells and smooth-muscle cells, particularly under conditions of injury repair or vascular lesion formation. Indeed, the ability of bone marrow to repair an injured endothelial monolayer may contribute to maintenance of vascular health, whereas failure to do so may lead to arterial disease. The precise sources of endothelial and mesenchymal progenitor cells or their stem cell precursors remain the subject of active investigation.
VASCULAR CELL BIOLOGY
Endothelial cell
The key cell of the vascular intima, the endothelial cell, has manifold functions in health and disease. Most obviously, the endothelium forms the interface between tissues and the blood compartment. It therefore must regulate the entry of molecules and cells into tissues in a selective manner. The ability of endothelial cells to serve as a selectively permeable barrier fails in many vascular disorders, including atherosclerosis and hypertension. This dysregulation of permeability also occurs in pulmonary edema and other situations of “capillary leak.”
The endothelium also participates in the local regulation of blood flow and vascular caliber. Endogenous substances produced by endothelial cells such as prostacyclin, endothelium-derived hyperpolarizing factor, nitric oxide (NO), and hydrogen peroxide (H2O2) provide tonic vasodilatory stimuli under physiologic conditions in vivo (Table 1-1). Impaired production or excess catabolism of NO impairs this endothelium-dependent vasodilator function and may contribute to excessive vasoconstriction in various pathologic situations. By contrast, endothelial cells also produce potent vasoconstrictor substances such as endothelin in a regulated fashion. Excessive production of reactive oxygen species, such as superoxide anion (O2–), by endothelial or smooth-muscle cells under pathologic conditions (e.g., excessive exposure to angiotensin II) can promote local oxidative stress and inactivate NO.
TABLE 1-1
ENDOTHELIAL FUNCTIONS IN HEALTH AND DISEASE
The endothelial monolayer contributes critically to inflammatory processes involved in normal host defenses and pathologic states. The normal endothelium resists prolonged contact with blood leukocytes; however, when activated by bacterial products such as endotoxin or proinflammatory cytokines released during infection or injury, endothelial cells express an array of leukocyte adhesion molecules that bind various classes of leukocytes. The endothelial cells appear to recruit selectively different classes of leukocytes in different pathologic conditions. The gamut of adhesion molecules and chemokines generated during acute bacterial infection tends to recruit granulocytes. In chronic inflammatory diseases such as tuberculosis and atherosclerosis, endothelial cells express adhesion molecules that favor the recruitment of mononuclear leukocytes that characteristically accumulate in these conditions.
The endothelium also dynamically regulates thrombosis and hemostasis. Nitric oxide, in addition to its vasodila-tory properties, can limit platelet activation and aggregation. Like NO, prostacyclin produced by endothelial cells under normal conditions not only provides a vasodilatory stimulus but also antagonizes platelet activation and aggregation. Thrombomodulin expressed on the surface of endothelial cells binds thrombin at low concentrations and inhibits coagulation through activation of the protein C pathway, inactivating clotting factors Va and VIIIa and thus combating thrombus formation. The surface of endothelial cells contains heparan sulfate glycosaminoglycans that furnish an endogenous antithrombotic coating to the vasculature. Endothelial cells also participate actively in fibrinolysis and its regulation. They express receptors for plasminogen and plasminogen activators and produce tissue-type plasminogen activators. Through local generation of plasmin, the normal endothelial monolayer can promote the lysis of nascent thrombi.
When activated by inflammatory cytokines, bacterial endotoxin, or angiotensin II, for example, endothelial cells can produce substantial quantities of the major inhibitor of fibrinolysis, plasminogen activator inhibitor 1 (PAI-1). Thus, in pathologic circumstances, the endothelial cell may promote local thrombus accumulation rather than combat it. Inflammatory stimuli also induce the expression of the potent procoagulant tissue factor, a contributor to disseminated intravascular coagulation in sepsis.
Endothelial cells also participate in the pathophysiology of a number of immune-mediated diseases. Lysis of endothelial cells mediated by complement provides an example of immunologically mediated tissue injury. The presentation of foreign histocompatibility complex antigens by endothelial cells in solid-organ allografts can trigger immunologic rejection. In addition, immune-mediated endothelial injury may contribute in some patients with thrombotic thrombocytopenic purpura and patients with hemolytic-uremic syndrome. Thus, in addition to contributing to innate immune responses, endothelial cells participate actively in both humoral and cellular limbs of the immune response.
Endothelial cells regulate growth of the subjacent smooth-muscle cells as well. Heparan sulfate glycosaminoglycans elaborated by endothelial cells can hold smooth-muscle proliferation in check. In contrast, when exposed to various injurious stimuli, endothelial cells can elaborate growth factors and chemoattractants, such as platelet-derived growth factor, that can promote the migration and proliferation of vascular smooth-muscle cells. Dysregulated elaboration of these growth-stimulatory molecules may promote smooth-muscle accumulation in atherosclerotic lesions.
Clinical assessment of endothelial function
Various invasive and noninvasive approaches can be used to evaluate endothelial vasodilator function in humans. Either pharmacologic agonists or increased flow stimulates the endothelium to release acutely molecular effectors that alter underlying smooth-muscle cell tone. Invasively, infusion of the cholinergic agonists acetylcholine and methacholine stimulates the release of NO from normal endothelial cells. Changes in coronary diameter can be quantitatively measured in response to an intracoronary infusion of these short-lived, rapidly acting agents. Noninvasive assessment of endothelial function in the forearm circulation typically involves occlusion of brachial artery blood flow with a blood pressure cuff, which elicits reactive hyperemia after release; the resulting flow increase normally causes endothelium-dependent vasodilation, which is measured as the change in brachial artery blood flow and diameter by ultrasound (Fig. 1-2). This approach depends on shear stress–dependent changes in endothelial release of NO after restoration of blood flow, as well as the effect of adenosine released (transiently) from ischemic tissue in the forearm.
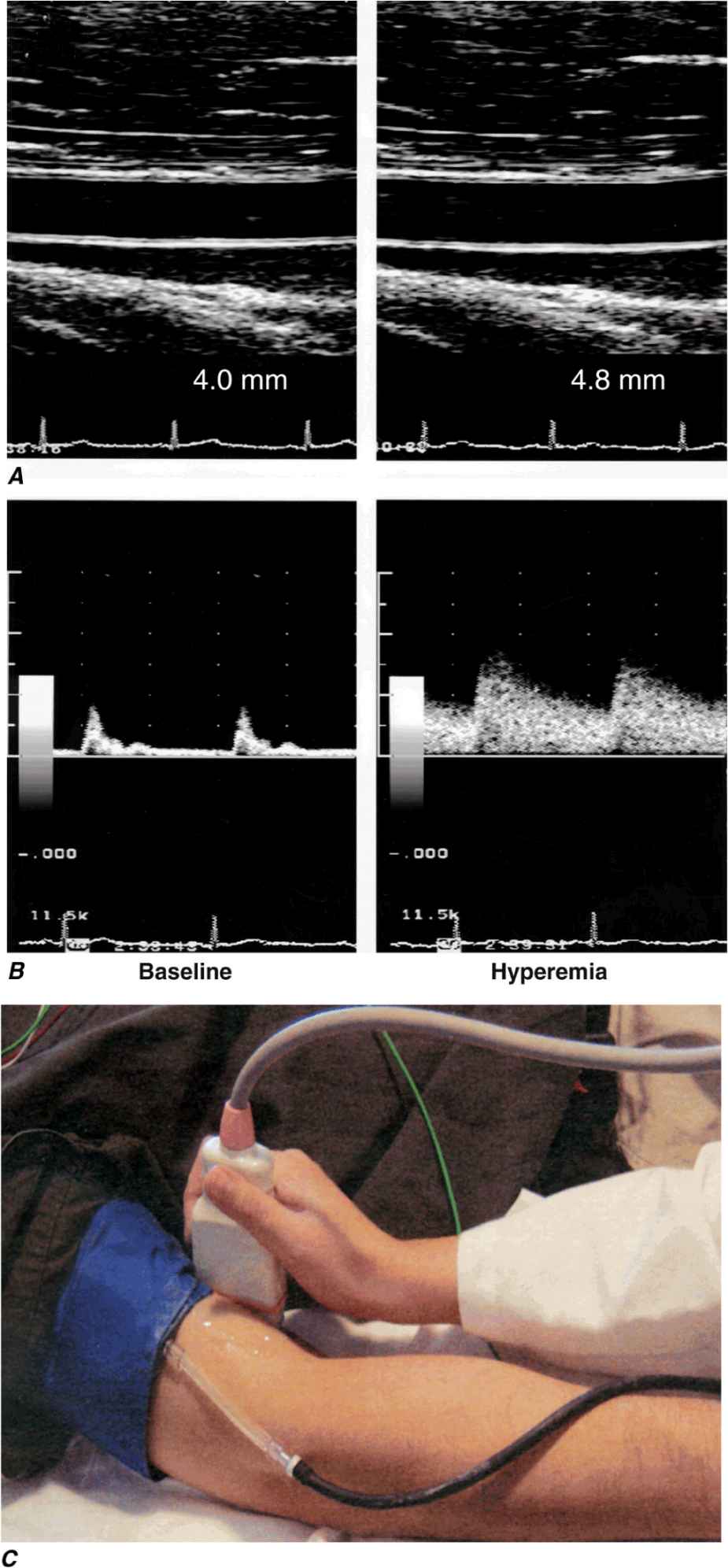
FIGURE 1-2
Assessment of endothelial function in vivo using blood pressure cuff-occlusion and release. Upon deflation of the cuff, changes in diameter (A) and blood flow (B) of the brachial artery are monitored with an ultrasound probe (C). (Reproduced with permission of J. Vita, MD.)
Typically, these invasive and noninvasive approaches detect inducible vasodilatory changes in vessel diameter of ~10%. In individuals with atherosclerosis or its risk factors (especially hypertension, hypercholesterolemia, diabetes mellitus, and smoking), such studies can detect endothelial dysfunction as defined by a smaller change in diameter and, in the extreme case, a so-called paradoxical vasoconstrictor response owing to the direct effect of cholinergic agonists on vascular smooth-muscle cell tone.
Vascular smooth-muscle cell
The vascular smooth-muscle cell, the major cell type of the media layer of blood vessels, also contributes actively to vascular pathobiology. Contraction and relaxation of smooth-muscle cells at the level of the muscular arteries controls blood pressure, and, hence, regional blood flow and the afterload experienced by the left ventricle (see later). The vasomotor tone of veins, which is governed by smooth-muscle cell tone, regulates the capacitance of the venous tree and influences the preload experienced by both ventricles. Smooth-muscle cells in the adult vessel seldom replicate. This homeostatic quiescence of smooth-muscle cells changes in conditions of arterial injury or inflammatory activation. Proliferation and migration of arterial smooth-muscle cells, which is associated with a change in phenotype characterized by lower content of contractile proteins and greater production of extracellular matrix macromolecules, can contribute to the development of arterial stenoses in atherosclerosis, arteriolar remodeling that can sustain and propagate hypertension, and the hyperplastic response of arteries injured by angioplasty or stent deployment. In the pulmonary circulation, smooth-muscle migration and proliferation contribute decisively to the pulmonary vascular disease that gradually occurs in response to sustained high-flow states such as left-to-right shunts. Such pulmonary vascular disease provides a major obstacle to the management of many patients with adult congenital heart disease. Elucidation of the signaling pathways that regulate the reversible transition of the vascular smooth-muscle cell phenotype remains an active focus of investigation. Among other mediators, microRNAs have emerged as powerful regulators of this transition, offering new targets for intervention.
The activated, phenotypically modulated smooth-muscle cells secrete the bulk of vascular extracellular matrix. Excessive production of collagen and glycosaminoglycans contributes to the remodeling and altered biology and biomechanics of arteries affected by hypertension or atherosclerosis. In larger elastic arteries, the elastin synthesized by smooth-muscle cells serves to maintain not only normal arterial structure but also hemodynamic function. The ability of the larger arteries, such as the aorta, to store the kinetic energy of systole promotes tissue perfusion during diastole. Arterial stiffness associated with aging or disease, as manifested by a widening pulse pressure, increases left ventricular afterload and portends a poor outcome.
Like endothelial cells, vascular smooth-muscle cells do not merely respond to vasomotor or inflammatory stimuli elaborated by other cell types but can themselves serve as a source of such stimuli. For example, when exposed to bacterial endotoxin or other proinflammatory stimuli, smooth-muscle cells can elaborate cytokines and other inflammatory mediators. Like endothelial cells, upon inflammatory activation, arterial smooth-muscle cells can produce prothrombotic mediators such as tissue factor, the antifibrinolytic protein PAI-1, and other molecules that modulate thrombosis and fibrinolysis. Smooth-muscle cells also elaborate autocrine growth factors that can amplify hyperplastic responses to arterial injury.
Vascular smooth-muscle cell function
Vascular smooth-muscle cells govern vessel tone. Those cells contract when stimulated by a rise in intracellular calcium concentration by calcium influx through the plasma membrane and by calcium release from intracellular stores (Fig. 1-3). In vascular smooth-muscle cells, voltage-dependent L-type calcium channels open with membrane depolarization, which is regulated by energy-dependent ion pumps such as the Na+,K+-ATPase pump and ion channels such as the Ca2+-sensitive K+ channel. Local changes in intracellular calcium concentration, termed calcium sparks, result from the influx of calcium through the voltage-dependent calcium channel and are caused by the coordinated activation of a cluster of ryanodine-sensitive calcium release channels in the sarcoplasmic reticulum (see later). Calcium sparks directly augment intracellular calcium concentration and indirectly increase intracellular calcium concentration by activating chloride channels. In addition, calcium sparks reduce smooth-muscle contractility by activating large-conductance calcium-sensitive K+ channels, hyperpolarizing the cell membrane and thereby limiting further voltage-dependent increases in intracellular calcium.
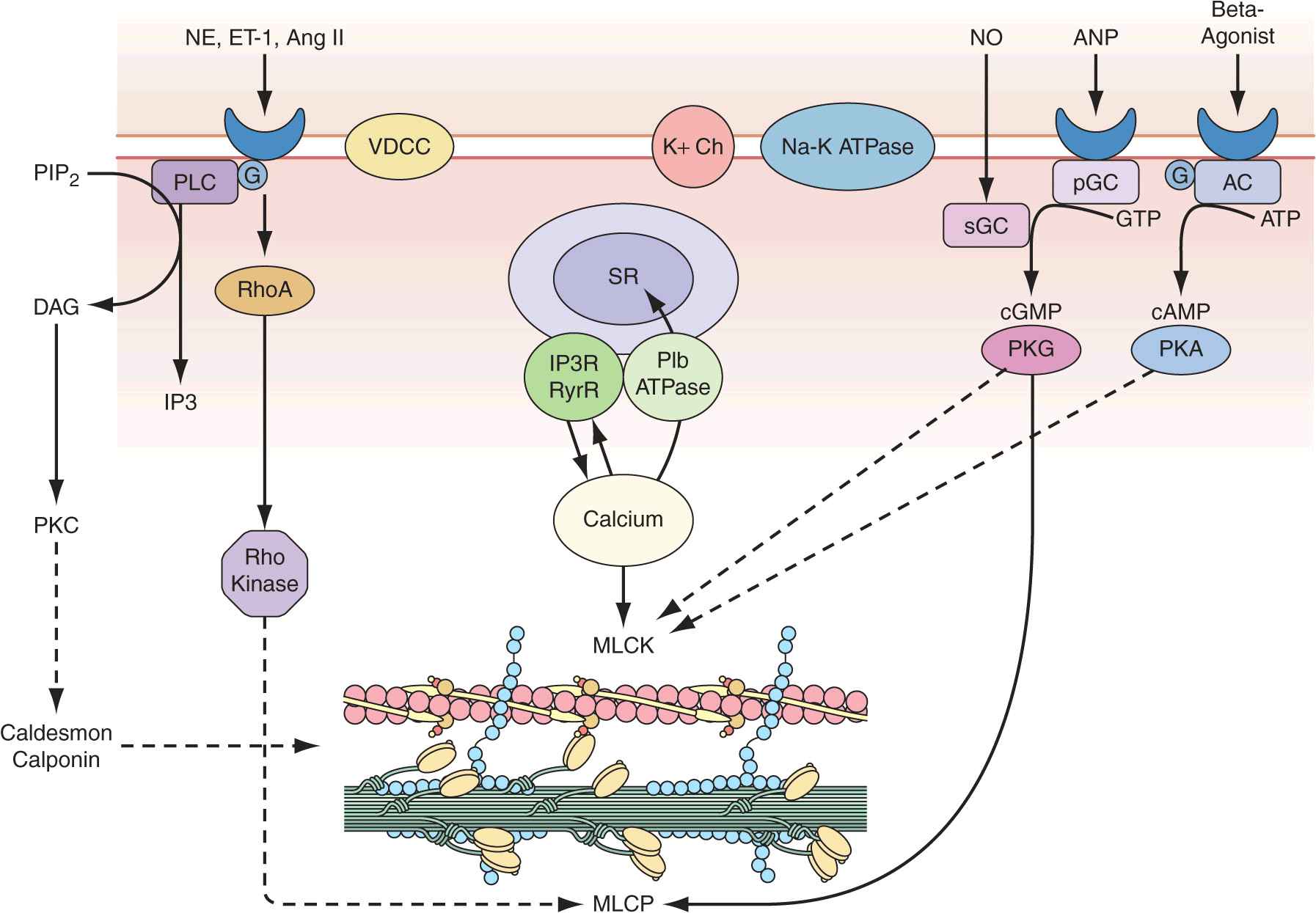
FIGURE 1-3
Regulation of vascular smooth-muscle cell calcium concentration and actomyosin ATPase-dependent contraction. AC, adenylyl cyclase; Ang II, angiotensin II; ANP, antrial natriuretic peptide; DAG, diacylglycerol; ET-1, endothelin-1; G, G-protein; IP3, inositol 1,4,5-trisphosphate; MLCK, myosin light chain kinase; MLCP, myosin light chain phosphatase; NE, norepinephrine; NO, nitric oxide; pGC, particular guanylyl cyclase; PIP2, phosphatidylinositol 4,5-bisphosphate; PKA, protein kinase A; PKC, protein kinase C; PKG, protein kinase G; PLC, phospholipase C; sGC, soluble guanylyl cyclase; SR, sarcoplasmic reticulum; VDCC, voltage-dependent calcium channel. (Modified from B Berk, in Vascular Medicine, 3rd ed, p 23. Philadelphia, Saunders, Elsevier, 2006; with permission.)
Biochemical agonists also increase intracellular calcium concentration, in this case by receptor-dependent activation of phospholipase C with hydrolysis of phosphatidylinositol 4,5-bisphosphate, resulting in generation of diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). These membrane lipid derivatives in turn activate protein kinase C and increase intracellular calcium concentration. In addition, IP3 binds to specific receptors on the sarcoplasmic reticulum membrane to increase calcium efflux from this calcium storage pool into the cytoplasm.
Vascular smooth-muscle cell contraction is controlled principally by the phosphorylation of myosin light chain, which in the steady state depends on the balance between the actions of myosin light chain kinase and myosin light chain phosphatase. Calcium activates myosin light chain kinase through the formation of a calcium-calmodulin complex. Phosphorylation of myosin light chain by this kinase augments myosin ATPase activity and enhances contraction. Myosin light chain phosphatase dephosphorylates myosin light chain, reducing myosin ATPase activity and contractile force. Phosphorylation of the myosin-binding subunit (thr695) of myosin light chain phosphatase by Rho kinase inhibits phosphatase activity and induces calcium sensitization of the contractile apparatus. Rho kinase is itself activated by the small GTPase RhoA, which is stimulated by guanosine exchange factors and inhibited by GTPase-activating proteins.
Both cyclic AMP and cyclic GMP relax vascular smooth-muscle cells through complex mechanisms. β agonists, acting through their G-protein-coupled receptors activate adenylyl cyclase to convert ATP to cyclic AMP; NO and atrial natriuretic peptide acting directly and via a G-protein-coupled receptor, respectively, activate guanylyl cyclase to convert GTP to cyclic GMP. These agents in turn activate protein kinase A and protein kinase G, respectively, which inactivate myosin light chain kinase and decrease vascular smooth-muscle cell tone. In addition, protein kinase G can interact directly with the myosin-binding substrate subunit of myosin light chain phosphatase, increasing phosphatase activity and decreasing vascular tone. Finally, several mechanisms drive NO-dependent, protein kinase G–mediated reductions in vascular smooth-muscle cell calcium concentration, including phosphorylation-dependent inactivation of RhoA; decreased IP3 formation; phosphorylation of the IP3 receptor–associated cyclic GMP kinase substrate, with subsequent inhibition of IP3 receptor function; phosphorylation of phospholamban, which increases calcium ATPase activity and sequestration of calcium in the sarcoplasmic reticulum; and protein kinase G–dependent stimulation of plasma membrane calcium ATPase activity, perhaps by activation of the Na+,K+-ATPase pump or hyperpolarization of the cell membrane by activation of calcium-dependent K+ channels.
Control of vascular smooth-muscle cell tone
The tone of vascular smooth-muscle cells is governed by the autonomic nervous system and by the endothelium in tightly regulated control networks. Autonomic neurons enter the blood vessel medial layer from the adventitia and modulate vascular smooth-muscle cell tone in response to baroreceptors and chemoreceptors within the aortic arch and carotid bodies and in response to thermoreceptors in the skin. These regulatory components include rapidly acting reflex arcs modulated by central inputs that respond to sensory inputs (olfactory, visual, auditory, and tactile) as well as emotional stimuli. Three classes of nerves mediate autonomic regulation of vascular tone: sympathetic, whose principal neurotransmitters are epinephrine and norepinephrine; parasympathetic, whose principal neurotransmitter is acetylcholine; and nonadrenergic/noncholinergic, which include two subgroups—nitrergic, whose principal neurotransmitter is NO, and peptidergic, whose principal neurotransmitters are substance P, vasoactive intestinal peptide, calcitonin gene-related peptide, and ATP.
Each of these neurotransmitters acts through specific receptors on the vascular smooth-muscle cell to modulate intracellular calcium and, consequently, contractile tone. Norepinephrine activates α receptors, and epinephrine activates α and β receptors (adrenergic receptors); in most blood vessels, norepinephrine activates postjunctional α1 receptors in large arteries and α2 receptors in small arteries and arterioles, leading to vasoconstriction. Most blood vessels express β2-adrenergic receptors on their vascular smooth-muscle cells and respond to β agonists by cyclic AMP–dependent relaxation. Acetylcholine released from parasympathetic neurons binds to muscarinic receptors (of which there are five subtypes, M1–5) on vascular smooth-muscle cells to yield vasorelaxation. In addition, NO stimulates presynaptic neurons to release acetylcholine, which can stimulate the release of NO from the endothelium. Nitrergic neurons release NO produced by neuronal NO synthase, which causes vascular smooth-muscle cell relaxation via the cyclic GMP–dependent and –independent mechanisms described earlier. The peptidergic neurotransmitters all potently vasodilate, acting either directly or through endothelium-dependent NO release to decrease vascular smooth-muscle cell tone.
The endothelium modulates vascular smooth-muscle tone by the direct release of several effectors, including NO, prostacyclin, hydrogen sulfide, and endothelium-derived hyperpolarizing factor, all of which cause vasorelaxation, and endothelin, which causes vasoconstriction. The release of these endothelial effectors of vascular smooth-muscle cell tone is stimulated by mechanical (shear stress, cyclic strain, etc.) and biochemical mediators (purinergic agonists, muscarinic agonists, peptidergic agonists), with the biochemical mediators acting through endothelial receptors specific to each class. In addition to these local paracrine modulators of vascular smooth-muscle cell tone, circulating mediators can affect tone, including norepinephrine and epinephrine, vasopressin, angiotensin II, bradykinin, and the natriuretic peptides (ANP, BNP, CNP, and DNP), as discussed earlier.
VASCULAR REGENERATION
Growth of new blood vessels can occur in response to conditions such as chronic hypoxemia and tissue ischemia. Growth factors, including vascular endothelial growth factor (VEGF) and forms of fibroblast growth factor (FGF), activate a signaling cascade that stimulates endothelial proliferation and tube formation, defined as angiogenesis. The development of collateral vascular networks in the ischemic myocardium reflects this process and can result from selective activation of endothelial progenitor cells, which may reside in the blood vessel wall or home to the ischemic tissue subtended by an occluded or severely stenotic vessel from the bone marrow. True arteriogenesis, or the development of a new blood vessel that includes all three cell layers, normally does not occur in the cardiovascular system of adult mammals. The molecular mechanisms and progenitor cells that can recapitulate blood vessel development de novo are under rapidly advancing study.
VASCULAR PHARMACOGENOMICS
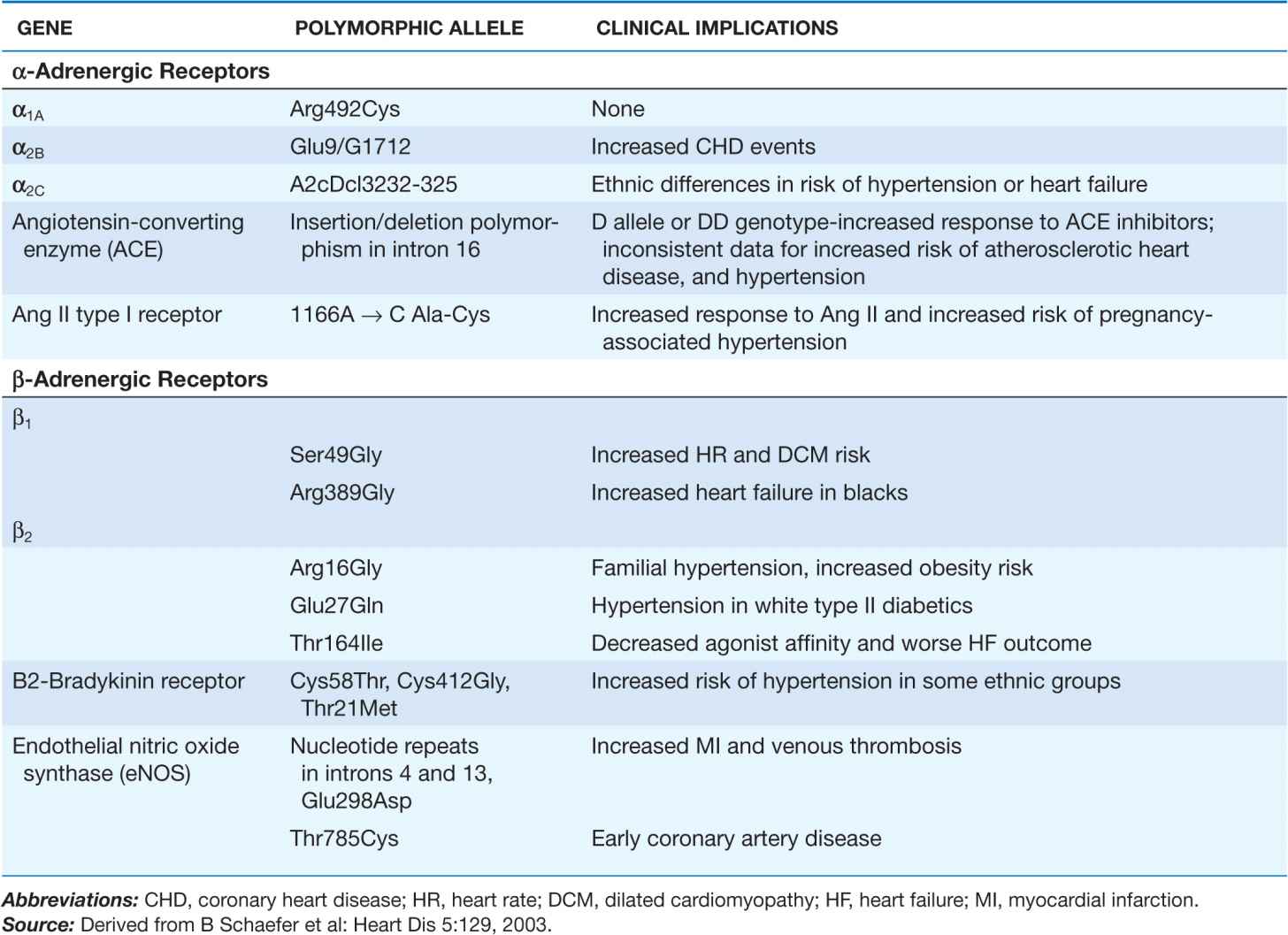
The last decade has witnessed considerable progress in efforts to define the genetic differences underlying individual variations in vascular pharmacologic responses. Many investigators have focused on receptors and enzymes associated with neurohumoral modulation of vascular function as well as hepatic enzymes that metabolize drugs that affect vascular tone. The genetic polymorphisms thus far associated with differences in vascular response often (but not invariably) relate to functional differences in the activity or expression of the receptor or enzyme of interest. Some of these polymorphisms appear to have different allele frequencies in specific ethnic groups. A summary of recently identified polymorphisms defining these vascular pharmacogenomic differences is provided in Table 1-2.
TABLE 1-2
GENETIC POLYMORPHISMS IN VASCULAR FUNCTION AND DISEASE RISK
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


