CHAPTER 116 Atrioventricular Canal Defects
Atrioventricular (AV) canal defects include a spectrum of lesions in which the common etiology appears to be abnormal development of the endocardial cushions, resulting in a defect in the AV septum and AV valves. This group of lesions forms approximately 3% of all major congenital cardiac defects, and approximately half of the patients have Down syndrome.1 In children with Down syndrome, AV canal defects are seen in 20% to 25%—a 1000-fold increased risk when compared with the incidence in the general population.2
Although AV canal defects constitute a continuum of related anatomic lesions, it is useful to divide them into two main groups3—partial and complete AV canal defects—on the basis of the extent of the interventricular communication:
ANATOMY
The anatomic malformation that is common to all forms of AV canal defects is a deficiency in the AV septum caused by incomplete embryonic development of superior and inferior endocardial cushion tissue. The common AV canal is normally present during the early tubular stage of fetal life and constitutes the sole connection between the primitive common atrium and the primitive common ventricle. After cardiac looping, the valves of the heart develop in the embryo from precursor structures, called endocardial cushions. These endocardial swellings become populated by valve precursor cells formed by a transformation from endothelial to mesenchymal tissue, and they undergo directed growth and remodeling to form the valvular structures and the membranous septa of the mature heart. (Fig. 116-1). Abnormal differentiation and remodeling of the cushion mesenchyme into valvuloseptal tissue is thought to be a mechanism for the development of AV canal defects.4 The resultant anatomic defect involves the abnormal development of AV valves and the persistence of interatrial and interventricular communications.5
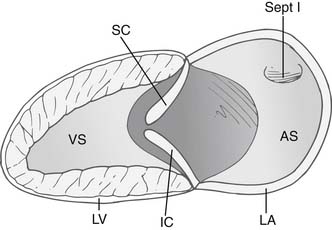
The AV valve in a canal defect has an area of apposition of leaflets that, unlike a normal commissure, is not supported by a papillary muscle. This area of apposition is called a cleft, and it is the apposition of the superior and inferior bridging leaflets. This cleft is not a commissure,6 as was once thought,7–9 for two reasons. First, a commissure is generally supported by chordae on either side of the defect, whereas a cleft is unsupported, with paucity of chordae at the edges. Second, the chordae that arise from the two adjacent leaflets in a commissure usually attach to a single papillary muscle, which promotes coaptation and prevents regurgitation. The chordae that arise from the left superior and inferior leaflets attach to two different papillary muscles. This increases the distracting force during ventricular systole and predisposes to regurgitation through the cleft. The extent of regurgitation is generally mild to moderate, although severe regurgitation can be present.
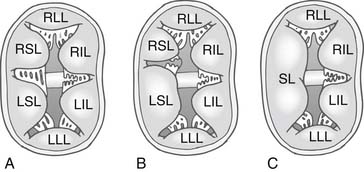
In 1966, Rastelli and colleagues10 described a classification of complete AV canal defect based on the extent of bridging of the left superior leaflet (LSL) across the interventricular septum (Fig. 116-2). In complete AV canal defects, the left inferior leaflet (LIL) is not well developed; it is usually short and immobile, with rolled edges. The Rastelli classification does not take into consideration the LIL, which displays greater anatomic variation. No clear morphologic relationship exists between the LSL and the LIL.
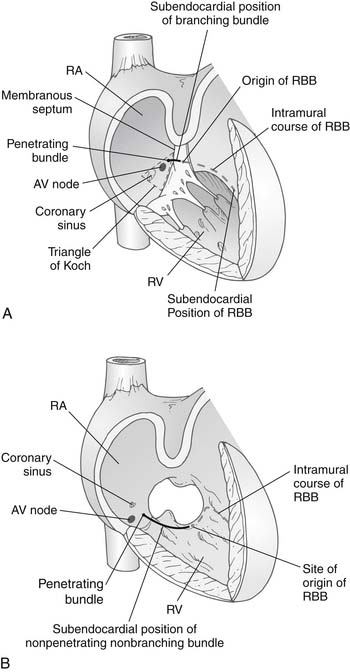
Lev’s11 description of the location of the conduction tissue was another important contribution in the field, because it facilitated the development of surgical techniques that avoided injury to the His bundle and the AV node. The coronary sinus ostium and the AV node are displaced inferiorly in AV canal defects. The AV node lies at the junction of the interatrial septum and the hinge point of the AV valve. This places the AV node between the coronary sinus ostium and the crest of the interventricular septum, in the so-called nodal triangle, which is not at the tip of Koch’s triangle. The nodal triangle is bound by the inferior extent of the right AV valve anulus, the coronary sinus orifice, and the inferior edge of the interatrial septum (Fig. 116-3). The bundle of His then passes superiorly and anteriorly from the AV node to the crest of the interventricular septum, reaching it where the crest is fused posteriorly with the AV valve anulus. The bundle then travels along the crest of the interventricular septum, in an inferior-to-superior direction, giving rise to the left bundle branches. Before reaching the midpoint of the interventricular crest, it becomes the right bundle branch and heads toward the moderator band and the muscle of Lancisi.12 The superior aspect of the interventricular septum is devoid of conduction fibers. The only exception is that, in patients with complete AV canal defects and heterotaxy syndrome, two AV nodes may be found.13
Approximately 50% to 75% of patients with complete AV canal have Down syndrome, whereas Down syndrome is rare in patients with partial AV canal defects,1 occurring in less than 10% of these patients. AV canal defects make up approximately 3% of all major congenital cardiac defects and are seen in approximately 25% to 30% of patients with Down syndrome. This represents a 1000-fold higher rate than in the general population.2 AV canal defects are the most common congenital heart anomaly in patients with Down syndrome. The presence of Down syndrome is thought to accelerate the development of pulmonary vascular obstructive disease (PVOD), especially in the setting of complete AV canal. An additional 15% to 20% of fetuses with AV canal syndrome have heterotaxy syndrome: AV canal defects are present in almost all patients with asplenia and in a high number of patients with polysplenia. Although an underlying genetic mechanism has not been identified, some intriguing evidence exists in that regard. First, there is a strong association between AV canal defects and Down syndrome. High levels of endostatin, which is known to regulate endocardial cushion development, have been found in patients with Down syndrome. Second, a weaker association exists with heterotaxy syndrome. Third, familial clustering of AV canal defects has been reported.14 Fourth, the incidence of recurrent congenital heart disease in offspring of patients with AV canal defects is approximately 10%.15 In another study, approximately 14% of offspring of mothers with isolated AV canal defects had congenital heart disease, usually either TOF or AV canal defects.16 This contrasts with the 2% to 4% probability of congenital heart disease in children of parents with other congenital cardiac lesions.
PATHOPHYSIOLOGY
In partial AV canal defects, where an interventricular communication is absent, the shunt is located only at the atrial level. Patients with partial AV canal defects with little or no regurgitation through the AV valves have a clinical presentation similar to that of patients with secundum ASDs. The degree of shunting depends on the size of the ostium primum ASD defect and the relative diastolic compliance of the two ventricles. RV stroke volume is increased, whereas RV systolic pressure may be normal or only slightly increased. Patients may remain asymptomatic for years. An exception is the presence of significant left AV valve regurgitation in the setting of an ostium primum ASD, which is the case in 10% to 15% of patients. The regurgitation increases left-to-right shunting considerably; a left-ventricle-to-right-atrium shunt may also be present. This results in increased RV and LV stroke volume, cardiomegaly, tachypnea, tachycardia, poor feeding, and failure to thrive, leading to progressive heart failure in infancy. Without treatment, patients with partial AV canal defects and large interatrial communications rarely survive beyond 40 years of age, although older patients have been reported.17 Atrial arrhythmias are common, increase in frequency with advancing age, and are a poor prognostic sign.
In many patients, particularly those with Down syndrome, pulmonary vascular resistance remains elevated after birth, which decreases the magnitude of left-to-right shunting. In other infants, pulmonary vascular resistance drops in the first few weeks of life, resulting in a large left-to-right shunt. Patients typically display tachypnea, failure to thrive, cardiomegaly, and diminished distal perfusion in the first few months of life. If AV valve regurgitation is coexistent, the extent of left-to-right shunting is increased because of shunting at both atrial and ventricular levels, resulting in biventricular volume overload. This accelerates the development of heart failure. In one study, moderate AV valve regurgitation was present in approximately 20% of complete AV canal defects, whereas severe regurgitation was present in 15% of such lesions.18 In our experience,19 mild AV valve regurgitation is common, occurring in approximately 35% of patients in the first year of life, although the incidence of severe AV valve regurgitation is less frequent, occurring in approximately 4% of patients.
Irreversible changes in the pulmonary vasculature are seen as early as 6 months in patients with unrepaired complete AV canal defects.20 The presence of Down syndrome accelerates the development of PVOD.21 This may result partly from an underlying pulmonary vascular defect predisposing to PVOD and partly from increased bronchial secretions, decreased number of distal bronchi, inherent abnormalities of the lung parenchyma, and the thick tongue that predisposes to oropharyngeal airway obstruction, hypoventilation, sleep apnea, tracheobronchomalacia, and CO2 retention. These factors accelerate the development of respiratory infections and PVOD. If PVOD progresses, eventually the shunt reverses to a right-to-left shunt and the Eisenmenger complex develops, with progressive cyanosis in later stages.
Without surgery, patients with complete AV canal defects and Down syndrome have an 80% survival rate at 10 and 15 years.22 The primary causes of death within the first several years of life are heart failure and recurrent pulmonary infections. Subsequently, PVOD becomes an increasingly prevalent cause of death. Toward the end of the second decade of life, exercise tolerance begins to decrease progressively, leading to premature death from PVOD by the third to fourth decades of life. One of the more dramatic causes of death in patients with PVOD is massive hemoptysis, which can occur as early as the third decade of life.
TIMING OF SURGERY
The first cardiac operation using a pump oxygenator was attempted by Dennis and Varco in 1952 in a patient with a preoperative diagnosis of an ASD. The patient did not survive surgery. Postmortem examination revealed a partial AV canal defect. In 1955, Lillehei and colleagues23 were the first to report repair of complete AV canal using cross-circulation. Successful repair of an ostium primum defect using cardiopulmonary bypass was first reported by Kirklin and colleagues in 1955.24
Complete Atrioventricular Canal
Two techniques have been proposed for repair of complete AV canal defects. A single patch covering atrial and ventricular defects with partition of the superior bridging leaflet was originally developed by Rastelli and Kirklin at the Mayo Clinic.25,26 Separate atrial and ventricular patches were proposed by Carpenter8 and advocated by others.27,28 A recent modification of the single-patch technique has been introduced.29,30
Management of the cleft has evolved over the years. Carpentier,8 Anderson and associates,7 and others9 recommended that the cleft be viewed as a commissure. They advocated leaving the cleft open to result in a “trifoliate” left AV valve and prevent the development of stenosis. However, the left-sided cleft is not a normal commissure, as discussed earlier. Left AV valve regurgitation frequently originates at the cleft; in later stages, as the regurgitation progresses, a central regurgitant jet may also be seen. Many surgeons in the 1980s left the cleft open only to have a sizable number of patients return with significant mitral regurgitation (MR) primarily through the cleft. For this reason, most surgeons today close the cleft completely. However, the cleft should be left open or only partially closed in patients with a single papillary muscle in the left ventricle, the so called parachute mitral valve, because mitral stenosis can result when complete cleft closure is undertaken in this setting.
We advocate repair in symptomatic infants regardless of age, and elective repair within the first 2 to 4 months of age in asymptomatic infants with complete AV canal defects. Delay beyond this age is unnecessary and is potentially hazardous. Repair of AV canal defects beyond 6 months of age has recently been shown to be an incremental risk factor for death.31 Freedom from reoperation for left AV valve regurgitation is higher when patients are repaired at a younger age.31 Prematurity and severe AV valve regurgitation should not be contraindications for corrective surgery, because the results with palliation in these groups are particularly poor.
Partial Atrioventricular Canal or Ostium Primum Defects
Sommerville32 reviewed the outcome of 122 unoperated patients with ostium primum ASDs between 1958 and 1964. Increasing morbidity and mortality occurred with increasing age. The presence of significant MR correlated with increasing symptoms. Of 96 patients younger than 30 years of age, 14% had died (n = 5) or had significant disabling symptoms (n = 8).
A subgroup of patients with partial AV canal defects display heart failure symptoms that are unresponsive to medical management in the first year of life.33 They require early surgical correction. Frequent associated findings include the presence of several left-sided obstructive lesions such as hypoplasia of the left AV valve, the left ventricle, the aortic valve, and the aortic arch, in addition to aortic coarctation. Complex mitral valve anomalies have also been documented in this subset of patients.34
PREOPERATIVE EVALUATION
In nearly all cases, the anatomic features, associated defects, and mechanism of valve dysfunction can be obtained from two-dimensional (2D) echocardiography, along with Doppler evaluation of blood flow direction and velocity.35 The four-chamber view shows the left and right AV valves at the same horizontal level, which is in contradistinction to the normal situation, in which the tricuspid valve is attached to the AV septum more toward the apex of the heart than the mitral valve. Other features on echocardiography include the elongated LV outflow tract and the unwedged aortic valve. It is important to assess the presence of chordal attachments to the LV outflow tract, because this is a substrate for the development of LVOTO. The AV valve leaflets should be examined, and the mechanism of AV valve regurgitation should be assessed carefully. The availability of real-time three-dimensional echocardiography (RT-3DE) has been very helpful in the assessment of the defect and to determine the mechanism of regurgitation. We use either epicardial or transesophageal RT-3DE in the assessment and management of AV valve repair, especially in reoperations for AV valve regurgitation. Studies have shown that RT-3DE can be successfully used in the repair of AV canal defects with a very short learning curve.35b
SURGICAL TECHNIQUE
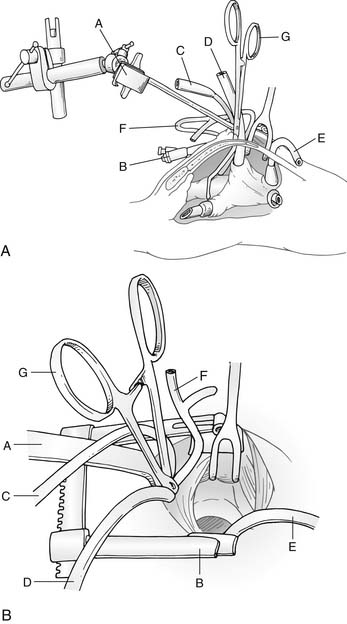
Median sternotomy, either partial or complete, has been the standard technique for exposing the heart and great vessels. Some have advocated a right anterior thoracotomy for partial AV canal defects in older female children to improve cosmesis. More recently, however, techniques to minimize surgical trauma by limiting the extent of the sternotomy have been used, such as a lower mini-sternotomy (Fig. 116-4).36 This approach can be applied to infants and children of all ages and may be helpful in decreasing postoperative pain and development of chest wall deformities such as pectus carinatum. We are now using this mini-sternotomy approach for nearly all AV canal defects, except when there are major associated cardiac lesions such as aortic coarctation, TOF, or TGA, where a full sternotomy is required for optimal exposure.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


