Atrial Fibrillation, Atrial Flutter, and Atrial Tachycardia: Introduction
Atrial fibrillation, atrial flutter, and atrial tachycardia are common arrhythmias associated with a variety of cardiac conditions. Atrial fibrillation, the most common sustained cardiac arrhythmia encountered in clinical practice, is increasing in prevalence.1-3 These arrhythmias may be associated with deterioration of hemodynamics, a wide spectrum of symptoms, and significant morbidity or mortality. Perhaps because no single therapy has been shown to be ideal for all patients, there are a variety of treatment strategies that may be applied to these arrhythmias. These include no therapy at all, anticoagulation, rhythm control, and rate control.3 This chapter describes the epidemiology, electrophysiologic mechanisms, and approach to management of patients with atrial fibrillation, atrial flutter, and atrial tachycardia.
Atrial Fibrillation
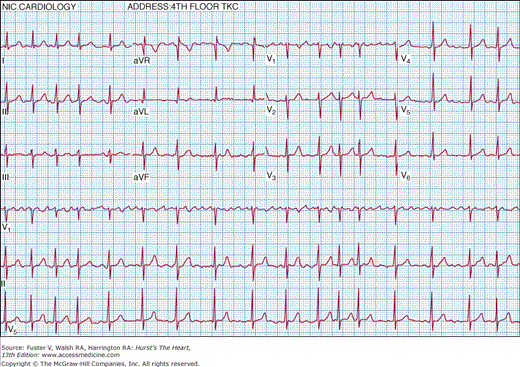
Atrial fibrillation (AF) is characterized by temporally and spatially varying rapid disorganized atrial electrical activation and uncoordinated atrial contraction. The surface electrocardiogram characteristically demonstrates rapid atrial fibrillatory waves with changing morphology and rate and a ventricular response that is usually irregularly irregular (Fig. 40–1)
The American Heart Association (AHA), American College of Cardiology (ACC), and the European Society of Cardiology (ESC) have proposed a standardized classification of AF into three categories: paroxysmal, persistent, and permanent.3 Paroxysmal AF is characterized by self-terminating episodes that generally last <7 days (most <24 hours), whereas persistent AF generally lasts >7 days and often requires electrical or pharmacologic cardioversion. AF is classified as permanent when it has failed cardioversion or when further attempts to terminate the arrhythmia are deemed futile. At the initial detection of AF, it may be difficult to be certain of the subsequent pattern of duration and frequency of recurrences. Thus a designation of first detected episode of AF is made on the initial diagnosis. When the patient has experienced ≥2 episodes, AF is classified as recurrent. The term lone AF refers to AF occurring in the absence of cardiac disease or other known etiologic factors, usually in relatively young individuals (generally <60 years of age).3 Most cases of AF occur in patients with evidence of structural heart disease, but there may be no evidence of concomitant disease in >50% of patients with paroxysmal AF.4-6 In contrast, >80% of patients with permanent AF have an identifiable underlying cause.7 The definition of chronic AF varies greatly in the literature and the terminology may be best avoided.
It is estimated that 2.2 to 5.0 million Americans and 4.5 million Europeans experience AF.1,3,8-12 The overall prevalence in the general population is estimated to be 0.4%.13 The incidence and prevalence of AF steadily increase with age, such that this arrhythmia occurs in <0.5% of the population <50 years of age and increases to approximately 2% at ages 60 to 69 years, 4.6% for ages 70 to 79 years, and 8.8% for ages 80 to 89 years.9,14,15 The age-adjusted prevalence of AF is higher for men than women9,10 and higher for whites than blacks.11 Familial AF has been described.3 In most cases, the genetic abnormality is unknown. However, specific genetic defects have been identified in a few families.16-19 In several Chinese families, the defect has resulted in a gain in function of potassium channels and shortening of atrial refractoriness.18,19
AF is associated with a wide variety of predisposing factors (Table 40–1). In the developed world, the most common clinical diagnoses associated with AF are hypertension and coronary artery disease.11 The presence of congestive heart failure (CHF) markedly increases the risk of AF.9-11 In developing countries, hypertension, rheumatic valvular heart disease, and congenital heart disease are the most commonly related conditions.20,21
| Diseases | Anatomic | Cellular | Electrophysio-Logic | |||
|---|---|---|---|---|---|---|
| Part A. Substrate develops during sinus rhythm (remodeling related to stretch and dilatation). The main pathways involve the RAAS, TGF-a, and CTGF. | ||||||
| Hypertension | Atrial dilatation | Myolysis | Conduction abnormalities | |||
| Heart failure | PV dilatation | Apoptosis, necrosis | ERP dispersion | |||
| Coronary disease | Fibrosis | Channel expression change | Ectopic activity | |||
| Valvular disease | ||||||
| Part B. Substrate develops because of tachycardia (tachycardia-related remodeling, downregulation of calcium channel, and calcium handling). | ||||||
| Focal AF | None orb | None orb | Ectopic activity | |||
| Atrial flutter | Atrial dilatation PV dilatation | Calcium channel downregulation | Microentry Short ERPc | |||
| Large PV sleeves | Myolysis | ERP dispersiond | ||||
| Reduced contractilitye | Connexin downregulation | Slowed conduction | ||||
| Fibrosis | Adrenergic supersensitivity | |||||
| Changed sympathetic innervation | ||||||
Different mechanisms may initiate (trigger) and maintain AF in an individual. The most frequent triggers are rapid spontaneous activity arising in the pulmonary veins. Pulmonary vein ectopy is usually transient, and persistence of AF after this mode of initiation may be by the multiple wavelet reentry mechanism (see later).
Spontaneous ectopy from muscular sleeves of pulmonary veins can serve as triggers of AF. Rapidly firing ectopic foci in pulmonary veins have been shown to be the underlying mechanism of most paroxysmal AF (Fig. 40–2).22-24 The pulmonary vein musculature of patients with paroxysmal AF demonstrates a markedly reduced effective refractory period and conduction delay.25 Rapidly firing foci can often be recorded within the pulmonary veins with conduction block to the left atrium (LA) (Fig. 40-3).23,24 Discontinuous properties of conduction within the pulmonary vein may also provide a substrate for reentry within the pulmonary vein itself26. Although most triggering foci that are mapped during electrophysiologic studies occur in the pulmonary veins in patients with paroxysmal AF, foci within the superior vena cava,27 the ligament of Marshall,28 and the musculature of the coronary sinus29 have been identified (Fig. 40–4). Other sites of initiating foci may be recorded in the left atrial wall or along the crista terminalis in the right atrium.30 For patients with pulmonary vein foci, a primary increase in adrenergic tone followed by a marked vagal predominance has been reported just before the onset of paroxysmal AF.31 A similar pattern of autonomic tone has been reported in an unselected group of patients with paroxysmal AF and a variety of cardiac conditions.32 Vagal stimulation shortens the refractory period of atrial myocardium but with a nonuniform distribution of effect. These factors support the importance of vagal stimulation in the induction of paroxysmal AF. In animal models, these pulmonary vein foci manifest delayed afterpotentials and triggered activity in response to catecholamine stimulation, rapid atrial pacing, or acute stretch.21,33
Figure 40–2
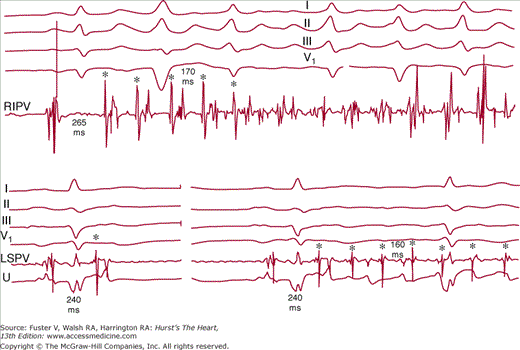
Top. Rapid firing in a pulmonary vein with the spontaneous onset of atrial fibrillation. Bottom left. Premature beats in a pulmonary vein (asterisk) with conduction block to the left atrium. Bottom right. Rapid firing at the same coupling interval conducts to the atrium with induction of atrial fibrillation. Reproduced with permission from Haïssaguerre et al.23
Figure 40–3

Simultaneous recordings of surface electrocardiographic lead I and bipolar electrograms from the right superior pulmonary vein (PV) of a patient after catheter ablation at the ostium of the vein produced conduction block from the vein into the left atrium. Note the rapid, irregular electrical activity recorded with a 64-electrode basket catheter within the pulmonary vein, with sinus rhythm in the remainder of the atria as recorded on the surface electrocardiogram.
Figure 40–4
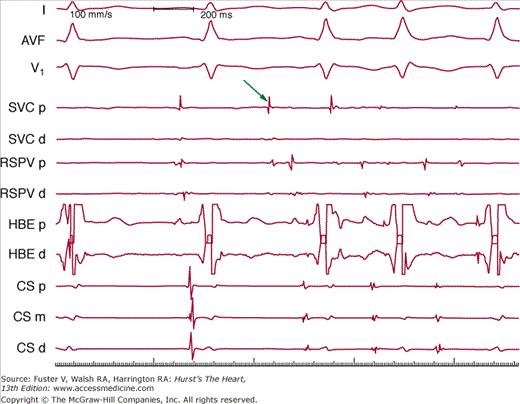
A rapidly firing focus in the superior vena cava (SVC) that induces atrial fibrillation. Surface leads I, aVF, and V1 are recorded simultaneously with intracardiac bipolar electrograms from the SVC proximal (SVC p) and distal (SVC d) pairs, right superior pulmonary vein (RSPV p and RSPV d), His bundle (HBE p and HBE d), and coronary sinus (CS p, CS m, CS d). The arrow indicates early activation in the superior vena cava.
A variety of electrophysiologic and structural factors promote the perpetuation of AF. Moe and colleagues34,35 proposed the multiple wavelet hypothesis as the mechanism of AF. Wavefronts traversing the atria fractionate into multiple daughter wavelets, the number of wavelets at any moment depends on the refractory period, conduction velocity, and anatomic obstacles in the atria. Li and colleagues36 demonstrated in a canine model of heart failure that interstitial fibrosis predisposed to intraatrial reentry and AF. Fibrosis of the atria may produce inhomogeneity of conduction within the atria, leading to conduction block and intraatrial reentry,37 a mechanism which has been shown in animal models.38,39 A variety of clinical studies have demonstrated that patients with AF have delayed interatrial conduction and inhomogeneous dispersion of atrial refractory periods.40 Long-standing AF results in loss of myofibrils, accumulation of glycogen granules, disruption in cell-to-cell coupling at gap junctions,41 and organelle aggregates.42,43 ADAMs (disintegrin and metalloproteinase), a family of membrane-bound glycoproteins that regulate cell–cell and cell–matrix interactions, have been reported to double in concentration during AF in human biopsies of atrial myocardium. This increased disintegrin and metalloproteinase activity may be one mechanism contributing to atrial dilatation in AF. Thus AF itself seems to produce a variety of alterations of atrial architecture that further contribute to atrial remodeling, mechanical dysfunction, and perpetuation of fibrillation.
In a population-based study of elderly patients without AF at baseline, Tsang and coworkers44 demonstrated that AF developed in direct relations to the echocardiographic left atrial volume index. An even stronger predictor of the development of nonvalvular AF was a restrictive transmitral Doppler flow pattern. Thus clinical evidence for diastolic dysfunction strongly supports the concept that myocardial stretch is an important mechanism of AF in the elderly. Altered stretch on atrial myocytes results in opening of stretch-activated channels.21 Force transmitted to stretch-activated channels in the membrane or via cytoskeletal integrins produces opening of these channels as well as increasing local production of angiotensin II, which in turn increases L-type Ca2+ current and decreases the transient outward K+ current. The antiarrhythmic effects of the angiotensin II receptor antagonist irbesartan have been demonstrated in patients undergoing electrical cardioversion.45 Stretch-activated channels increase G protein–coupled pathways, leading to increased protein kinase A and C activity and increased L-type Ca2+ current through the cell membrane and release of Ca2+ from the sarcoplasmic reticulum (promoting after depolarizations and triggered activity).21
AF also produces electrical remodeling that promotes further AF. The electrophysiologic changes typical of atrial myocytes during AF are a decrease in effective refractory period, decrease in action potential duration, reduction in the amplitude of the action potential plateau, and a loss of response of action potential duration to changes in rate (abnormal restitution).46 Although the normal atrial action potential duration shortens in response to pacing at shorter cycle lengths, AF results in loss of this rate dependence of the action potential.47-51 These changes can be attenuated by the sarcoplasmic reticulum’s release of the calcium antagonist ryanodine, suggesting the importance of increased intracellular calcium to the maladaptation of atrial myocardium during AF. The reduction in the atrial effective refractory periods produced by AF are not uniform throughout the atria.47 Most studies have demonstrated a reduction in atrial conduction velocity in response to prolonged rapid atrial rates.48-50 The presence of underlying CHF may further interact with these electrophysiologic changes to promote AF. For example, Shinagawa and colleagues47 reported that prolonged AF could be induced in 0 of 14 control dogs, 2 of 14 dogs with CHF, 4 of 12 dogs subjected to rapid atrial pacing at 400 beats/min for 1 week, and 8 of 13 dogs with both CHF and rapid atrial pacing. Similar data have been reported by Stambler and coworkers51 in a canine, rapid ventricular pacing, heart failure model; however, importantly, in this model, the atrial effective refractory period increases, and the response of the action potential (restitution) is preserved.51
Clinical presentation of AF may relate to the disease itself or its consequences (thromboembolism and tachycardia induced cardiomyopathy). Patients may exhibit none to disabling symptoms. Common symptoms include anxiety, palpitations, dyspnea, dizziness, chest pain, and fatigue. Several hemodynamic derangements, including rapid ventricular rates, loss of organized atrial contraction, irregularity of cardiac rhythm, and bradycardia (resulting particularly from sinus pauses when AF episodes terminate) may be the underlying cause of the symptoms. Many patients with symptomatic AF also have asymptomatic episodes.52,53 Several studies have reported a marked reduction in quality of life in patients with AF.54,55 The ACC/AHA/ESC task force on practice guidelines has recommended that an evaluation for patients with AF should include a history and physical examination, electrocardiogram, chest radiograph, echocardiogram, and thyroid function studies3 (Table 40–2).
| Minimum Evaluation | Additional Testing |
|---|---|
| 1. To define history and physical examination | One or several tests may be necessary |
| Presence and nature of symptoms associated with AF | 1. Six-minute walk test |
| Clinical type of AF (first episode, paroxysmal, persistent, or permanent) | If the adequacy of rate control is in question |
| Onset of the first symptomatic attack or date of discover of AF | 2. Exercise testing |
| Frequency, duration, precipitating factors, and modes of termination of AF | If the adequacy of rate control is in question (permanent AF) |
| Response to any pharmacologic agents that have been administered | To reproduce exercise-induced AF |
| Presence of any underlying heart disease or other reversible | To exclude ischemia before treatment of selected patients |
| conditions (eg, hyperthyroidism or alcohol consumption) | with a type 1C antiarrhythmic drug |
| 2. To identify electrocardiogram | 3. Holter monitoring or event recording |
| Rhythm (verify AF) | If diagnosis of the type of arrhythmia is in question |
| LV hypertrophy | As a means of evaluating rate control |
| P wave duration and morphology or fibrillatory waves | 4. Transesophageal echocardiography |
| Preexcitation | To identify LA thrombus (in the LA appendage) |
| Bundle-branch block | To guide cardioversion |
| Prior MI | 5. Electrophysiological study |
| Other atrial arrhythmias | To clarify the mechanism of wide-WRS-complex tachycardia |
| To measure and follow the R-R, QRS, and QT intervals in | To identify a predisposing arrhythmia such as atrial flutter or paroxysmal |
| conjunction with antiarrhythmic drug therapy | supraventricular tachycardia |
| 3. To identify transthoracic echocardiogram | To seek sites for curative ablation or AV conduction block/modification |
| Valvular heart disease | 6. To evaluate chest radiograph |
| LA and RA size | Lung parenchyma, when clinical findings suggest an abnormality |
| LV size and function | Pulmonary vasculature, when clinical findings suggest an abnormality |
| Peak RV pressure (pulmonary hypertension) | |
| LV hypertrophy | |
| LA thrombus (low sensitivity) | |
| Pericardial disease | |
| 4. Blood tests of thyroid, renal, and hepatic function | |
| For a first episode of AF, when the ventricular rate is difficult to control |
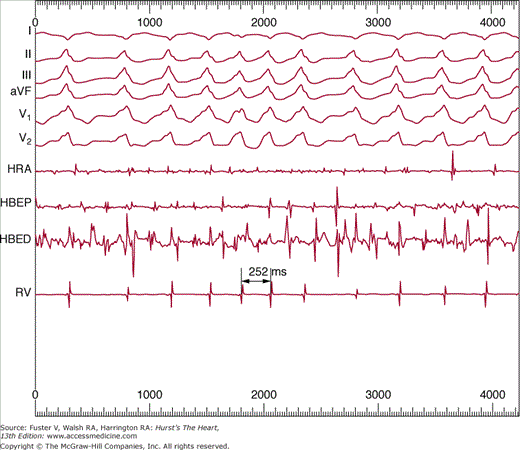
The ventricular rate during AF can be quite variable and depends on autonomic tone, the electrophysiologic properties of the atrioventricular (AV) node, and the effects of medications that act on the AV conduction system. The ventricular rate may be very rapid (>300 beats/min) in patients with the Wolff-Parkinson-White syndrome, with conduction over accessory pathways (wide preexcited QRS complexes) having short anterograde refractory periods (Fig. 40–5). A regular, slow ventricular response during AF suggests an AV junctional rhythm, either as an escape mechanism with complete AV block or as an accelerated AV junctional pacemaker.
Stroke is the most common thromboembolic event in AF and approximately 36% of all strokes in individuals aged 80 to 89 years are attributed to AF.56 Furthermore, strokes occurring in patients who have AF tend to have a higher degree of severity.57 Individuals who have AF are not at equal risk for thromboembolic events and several predisposing clinical factors can identify those patients at high risk (discussed later). Most thrombi associated with AF arise within the left atrial appendage.58 Flow velocity within the left atrial appendage is reduced during AF because of the loss of organized mechanical contraction.59 Compared with transthoracic echocardiogram, the transesophageal echocardiogram offers a much more sensitive and specific means of assessing left atrial thrombi and spontaneous echo contrast, an indicator of reduced flow.60 Several factors contribute to the enhanced thrombogenicity of AF. Nitric oxide (NO) production in the left atrial endocardium is reduced in experimental AF, with an increase in levels of the prothrombotic protein plasminogen activator inhibitor 1 (PAI-1).61 The lowest levels of NO and the highest levels of PAI-1 were recorded in the left atrial appendage during AF. Patients with AF have elevated levels of β-thromboglobulin and platelet factor 462,63; elevated plasma levels of von Willebrand factor (vWF), soluble thrombomodulin, and fibrinogen have been reported in patients with permanent AF with no evidence of diurnal variation in thrombogenicity.64,65 In the Stroke Prevention in Atrial Fibrillation (SPAF) III study,66 increased plasma levels of vWF were strongly correlated with the clinical predictors of stroke in AF (age, prior cerebral ischemia, CHF, diabetes, and body mass index). There was a stepwise increase in vWF with increasing clinical risk of stroke in this population.
In occasional patients, the first clinical manifestation of AF may be CHF related to a tachycardia-induced cardiomyopathy.67,68 The left ventricular (LV) dysfunction develops due to sustained high ventricular rates during AF. This clinical syndrome generally occurs in asymptomatic patients with a sustained high ventricular rates (usually >120 beats/min) for prolonged time periods. Because the patients do not experience symptoms, they do not seek medical care initially and often come to medical attention with signs and symptoms of heart failure. In these patients, control of the ventricular rate typically reverses the impaired LV function within weeks. The severity and temporal course of its onset varies significantly between individuals. In experimental models, ventricular dysfunction can occur as soon as 24 hours and continue to deteriorate for 3 to 5 weeks with rapid pacing rates. Recovery of ventricular function with cessation of pacing could start within 48 hours, and normalization can occur within 1 to 2 weeks.69
AF produces several adverse hemodynamic effects, including loss of atrial contraction, a rapid ventricular rate, and an irregular ventricular rhythm. The loss of mechanical AV synchrony may have a dramatic impact on ventricular filling and cardiac output when there is reduced ventricular compliance, as with LV hypertrophy from hypertension, restrictive cardiomyopathy, hypertrophic cardiomyopathy, or the increased ventricular stiffness associated with aging. In addition, patients with mitral stenosis, constrictive pericarditis, or right ventricular infarction typically experience marked hemodynamic deterioration at the onset of AF. The loss of AV synchrony results in a decrease in LV end-diastolic pressure (LVEDP) as the loading effect of atrial contraction is lost, thereby reducing stroke volume and LV contractility by the Frank-Starling mechanism. Although there is a reduction in the LVEDP, there is an increase in the left atrial mean diastolic pressure. Patients with significant restrictive physiology may experience pulmonary edema and/or hypotension with the onset of AF. In contrast, patients with dilated cardiomyopathy and high LV filling pressures may experience minimal hemodynamic compromise with AF if their LV compliance is not significantly impaired. The inappropriately rapid ventricular rate during AF also limits the duration of diastole and reduces ventricular filling. The irregular ventricular rhythm has adverse hemodynamic effects that are independent of the ventricular rate. Irregular rhythm may significantly reduce cardiac output70 and coronary blood flow71 compared with a regular ventricular rhythm at the same average heart rate. The effect of ventricular irregularity on coronary blood flow may explain in part why some patients with AF experience precordial pain in the presence of normal coronary arteriography.
Prevention of the disease-related complications (thromboembolism and tachycardia-induced cardiomyopathy) and control of symptoms may be considered the primary goals of AF management. The three major therapeutic strategies in managing AF include prevention of stroke, rate control, and rhythm control. Anticoagulation with warfarin reduces the risk for stroke. Therapies to achieve symptom control and prevention of tachycardia-induced cardiomyopathy are often similar. For example, ventricular rate control during AF or maintenance of sinus rhythm may improve symptoms and prevent tachycardia-induced cardiomyopathy. When clinical goals are not met using one strategy, the alternate strategy can be pursued in the same patient. Current therapies of AF have not demonstrated survival benefits, and future research needs to focus on the goals of improving the survival of patients with AF. Development of strategies for the primary prevention of AF is another area of great significance for research considering the high prevalence of the disease.
Not all AF patients are at high risk for stroke. The recognized clinical markers predicting increased risk for stroke in AF are prior stroke or transient ischemic attack, hypertension, diabetes mellitus, congestive heart failure, and age older than 75 years (Table 40–3).3 Other less validated stroke risk factors include coronary artery disease, thyrotoxicosis, female sex, LV dysfunction, and age older than 65 years.3 Mitral stenosis is well known to be associated with high risk for stroke in AF patients and warfarin anticoagulation is indicated in all such patients.
| CHADS2 Risk Criteria | Score | |
|---|---|---|
| Prior Stroke or TIA | 2 | |
| Age >75 y | 1 | |
| Hypertension | 1 | |
| Diabetes mellitus | 1 | |
| Congestive heart failure | 1 | |
| Patients (n = 1733) | Adjusted Stroke Rate (%/y)a (95% CI) | CHADS2 Score |
| 120 | 1.9 (1.2-3.0) | 0 |
| 463 | 2.8 (2.0-3.8) | 1 |
| 523 | 4.0 (3.1-5.1) | 2 |
| 337 | 5.9 (4.6-7.3) | 3 |
| 220 | 8.5 (6.3-11.1) | 4 |
| 65 | 12.5 (8.2-17.5) | 5 |
| 5 | 18.2 (10.5-27.4) | 6 |
The CHADS2 stroke risk stratification scheme, which is based on analysis of 1773 patients in the National Registry for Atrial Fibrillation, has gained considerable favor and is used in the ACC/AHA/ESC 2006 management guidelines to tailor therapy for stroke prevention.3,72 Each of the letters in this acronym represents a risk factor—Congestive heart failure, Hypertension, Age, Diabetes, and Stroke.72 Previous stroke or transient ischemic attack is the strongest predictor of stroke and therefore carries 2 points, whereas the other risk factors carry 1 point each. The adjusted stroke rate per 100 patient-years increases from 1.9 with a score of 1 to 18.2 with a score of 6 (see Table 40–3). The recommendations for antithrombotic therapy to prevent thromboembolism in patients with AF is summarized in Table 40–4.3 There is widespread consensus that all patients with rheumatic valvular heart disease and AF require anticoagulation with warfarin unless there is an absolute contraindication. There is no difference in the indications for antithrombotic therapy between paroxysmal, persistent, or permanent AF.
| Risk Category | Recommended Therapy | |
|---|---|---|
| No risk factors | Aspirin 81-325 mg daily | |
| One moderate-risk factor | Aspirin 81-325 mg daily or warfarin (INR 2.0-3.0, target 2.5) | |
| Any high-risk factor or >1 moderate-risk factor | Warfarin [INR 2.0-3.0, target 2.5]a | |
| Less Validated or Weaker Risk Factors | Moderate-Risk Factors | High-Risk Factors |
| Female sex | Age ≥75 y | Previous stroke, TIA, or embolism |
| Age 65-74 y | Hypertension | Mitral stenosis |
| Coronary artery disease | Heart failure | Prosthetic heart valve |
| LV ejection fraction ≤35% | ||
| Thyrotoxicosis | Diabetes mellitus |
Warfarin is remarkably effective at reducing stroke risk in patients with AF. This was clearly demonstrated by a meta-analysis of five randomized, controlled clinical trials comparing warfarin versus placebo by the AF Investigators:
Copenhagen Atrial Fibrillation Aspirin and Anticoagulation trial
SPAF trial
Boston Area Anticoagulation Trial for Atrial Fibrillation
Canadian Atrial Fibrillation trial
Stroke Prevention in Nonrheumatic Atrial Fibrillation trial (Fig. 40–6)
Figure 40–6
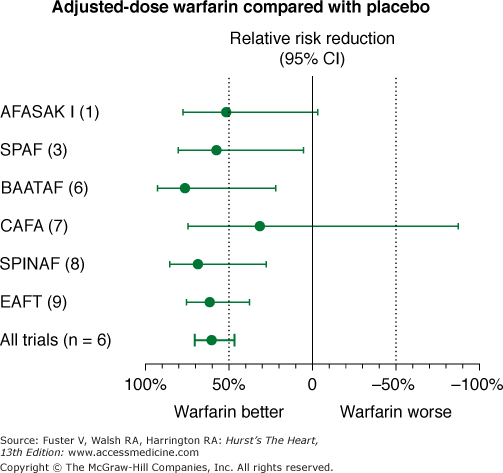
Effects of warfarin versus placebo on risk of stroke in six randomized, placebo-controlled clinical trials in nonvalvular atrial fibrillation. AFASAK I (1), The Copenhagen Atrial Fibrillation, Aspirin, and Anticoagulant Therapy Study; BAATAF (6), Boston Area Anticoagulation Trial for Atrial Fibrillation; CAFA (7), Canadian Atrial Fibrillation Anticoagulation; CI, confidence interval; EAFT (9), European Atrial Fibrillation Trial; SPAF (3), Stroke Prevention in Atrial Fibrillation; SPINAF (8), Stroke Prevention in Atrial Fibrillation. Data from Hart et al.75
Using an intention-to-treat analysis that compared warfarin therapy with placebo, there was a 68% risk reduction in stroke for patients taking warfarin compared with patients taking placebo (P < .001).73 Moreover, a subsequent on-treatment analysis demonstrated an 83% risk reduction in stroke when patients were taking warfarin compared with placebo.74 Warfarin should be administered to achieve an international normalized ratio (INR) between 2 and 3, for optimal efficacy and safety (Fig. 40–7).
Figure 40–7
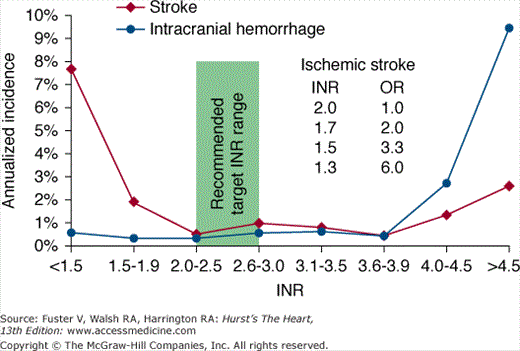
Annualized incidence of stroke or intracranial hemorrhage according to the international normalized ratio (INR). Note that when the INR decreases to <2.0, there is a steep rise in the odds ratio (OR) for stroke, but the incidence of intracranial hemorrhage remains low and flat (ie, there is no associated decrease in intracranial hemorrhage). Modified with permission from Hylek et al.78,79
Meta-analysis of studies comparing aspirin with placebo suggests a relative risk reduction of approximately 22% with aspirin (Fig. 40–8). However, this suggestion of benefit is largely driven by the SPAF I data.75 Comparisons of warfarin versus aspirin show superiority of warfarin (Fig. 40–9). In addition, the SPAF III trial76 evaluated the benefit of an adjusted dose of warfarin (INR 2-3; target 2.5) versus low-intensity, fixed-dose warfarin (INR 1.2-1.5) plus aspirin in patients with AF at high risk for stroke.77 The trial was stopped early because the event rate in patients on the combination therapy was 7.9% per year versus an event rate on adjusted dose warfarin of 1.9% per year (P = .001). Additional concerns using aspirin for stroke prevention in lieu of warfarin relate to severity of stroke.78,79 Thus these data found that warfarin with an INR ≥2 not only reduces the frequency of ischemic stroke, it also reduces the severity and risk of death from stroke compared with aspirin.
Figure 40–8
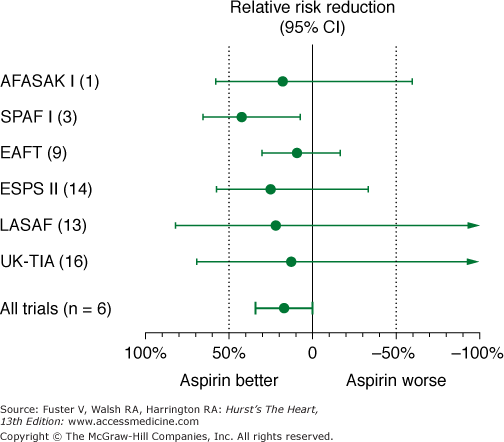
Effects of aspirin versus placebo on risk of stroke in 5 randomized, placebo-controlled trials in nonvalvular atrial fibrillation. AFASAK I (1), The Copenhagen Atrial Fibrillation, Aspirin, and Anticoagulant Therapy Study; CI, confidence interval; EAFT (9), European Atrial Fibrillation Trial; ESPS II (14), European Stroke Prevention Study; LASAF (13), Alternate-Day Dosing of Aspirin in Atrial Fibrillation Pilot Study Group; SPAF I (3), Stroke Prevention in Atrial Fibrillation; UK-TIA (16), United Kingdom Transient Ischaemic Attack Trial. Data from Hart et al.75
Figure 40–9
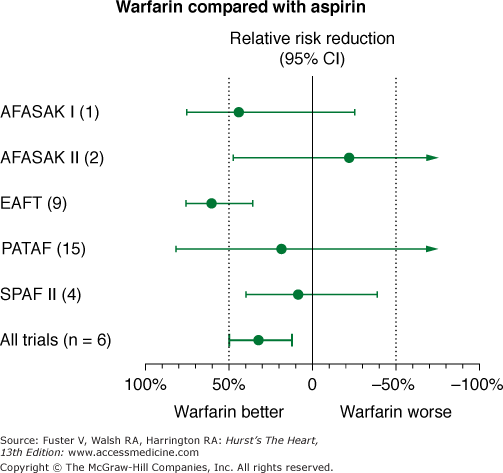
Effects of aspirin versus warfarin on risk of stroke in 5 randomized, controlled clinical trials in nonvalvular atrial fibrillation. AFASAK I (1) and AFASAK II (2), The Copenhagen Atrial Fibrillation, Aspirin, and Anticoagulant Therapy Study; EAFT (9), European Atrial Fibrillation Trial; PATAF (15), Primary Prevention of Arterial Thromboembolism in Non-rheumatic Atrial Fibrillation; SPAF II (4), Stroke Prevention in Atrial Fibrillation. Data from Hart et al.75
There have been several recent trials demonstrating potential alternative therapies to reduce risk of stroke in patients with AF.80-82 In a randomized clinical trial of patients with AF with an increased risk of stroke who were deemed unsuitable for warfarin, the combination of aspirin plus clopidogrel compared with aspirin alone had a small, but significant reduction in the risk of major vascular events, especially stroke, but also had an increased risk of major hemorrhage.80 Dabigatran, a direct competitive inhibitor of thrombin, was compared with warfarin in a randomized trial of patients with AF. At a dose of 110 mg twice daily, dabigatran was associated with rates of systemic embolism and stroke similar to those noted with warfarin, but at a dose of 150 mg twice daily compared with warfarin, dabigatran had a lower rate of stroke and systemic embolism, with similar rates of major hemorrhage.81 Lastly, preliminary data with a percutaneous closure device introduced into the left atrial appendage demonstrated noninferiority to warfarin therapy for the effectiveness of stroke prevention.82
In summary, it is important that the risks of bleeding versus the benefits of stroke prevention always be weighed for each patient. However, the risk of stroke typically is greater than the risk of bleeding for most patients with AF at substantial risk for stroke.
Several prospective, randomized trials have been published comparing the strategies of rate control and rhythm control in patients with AF.83-85 The AFFIRM trial enrolled 4060 patients aged older than 65 years or with risk factors for stroke, randomizing them to rate versus rhythm control.83 Over a mean follow-up period of 3.5 years, there was no significant difference in overall mortality between the two groups (Fig. 40–10). The RACE trial84 randomly assigned 522 patients with persistent AF after electrical cardioversion to either a rate-control or a rhythm-control treatment. There was no difference in the primary end point of the study (a composite of cardiovascular death, CHF, thromboemboli, bleeding, need for pacemaker, or serious drug side effects) between the two strategies. Thus these studies indicate that both approaches have similar clinical outcomes provided that appropriate anticoagulation is maintained.
Figure 40–10

Results of rate control versus rhythm control on overall survival in the Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) trial. Reproduced with permission from Wyse et al.83
In view of these randomized trials, how should an individual patient with AF be managed? The patients enrolled in these studies have been older and typically had ≥1 risk factors for stroke. The results concerning morbidity and mortality may not extend to other patient populations. Our bias is a rhythm-control strategy in younger patients and a rate-control strategy in older patients with AF, but in the end, this should be decided on a case-by-case basis. Anticoagulation is needed in patients at high risk for stroke regardless of whether a rate or rhythm-control strategy is chosen.
Control of the ventricular rate involves both acute and chronic phases. In the acute phase, intravenous metoprolol, esmolol, diltiazem, or verapamil have all been demonstrated to provide slowing of AV nodal conduction; these drugs are indicated for patients with severe symptoms related to a rapid ventricular rate.3 Intravenous digoxin requires a longer duration to achieve rate control and is less useful. For patients with only mild or moderate symptoms, oral medications that slow AV nodal conduction should be prescribed. After control of the resting ventricular rate has been achieved, attention is paid to the ambulatory heart rate. There is no overall agreement on what constitutes optimum rate control.86 One set of criteria is 60 to 80 beats/min at rest and between 90 and 115 beats/min during moderate exercise.3 It is also uncertain which method is best to evaluate rate control, which has been done using a resting 12 ECG, 24-hour Holter monitor, exercise testing, and combinations of methods.86 In essence, we want to achieve rate control for each patient during their usual daily activities, so we typically recommend titrating AV nodal blockers on an outpatient basis following the daily 24-hour heart rate plot as a guide.86
Digoxin may provide effective control of the resting heart rate but is often ineffective during exertion. β-Adrenergic blockers or calcium channel antagonists provide much better control of the ventricular rate during exercise and should be considered for most patients. Digoxin is most useful in the setting of impaired systolic function and can be used in combination with β-blockers or calcium antagonists if these agents do not provide adequate rate control. Control of the ventricular rate can be especially challenging for patients with the tachycardia-bradycardia syndrome, who experience rapid ventricular rates during AF and sinus bradycardia or sinus pauses when AF terminates. Permanent pacemaker implantation is indicated for many of these patients.
Some patients may continue to experience significant symptoms from a rapid or irregular ventricular rhythm despite drug therapy. Catheter ablation of the AV conduction system and permanent pacemaker implantation is a highly effective means of establishing permanent control of the ventricular rate during AF in selected patients.87-89 Despite the many favorable effects of this procedure, there are several limitations. First, AV nodal ablation does not change the long-term need for anticoagulation. Second, although an adequate junctional escape rhythm may be present after ablation, some patients may become pacemaker dependent due to an inadequate escape rhythm. Third, right ventricular pacing produces an abnormal LV contraction sequence, and acute worsening of hemodynamics has been observed in some patients. In the Post AV Node Ablation Evaluation (PAVE) trial, patients who received biventricular versus right ventricular apical pacing, especially those with abnormal LV ejection fractions before ablation, had longer 6-minute walking distances and higher LV ejection fractions after ablation.90
When a rhythm-control strategy is chosen for patients with paroxysmal or persistent AF, prophylactic treatment with antiarrhythmic drugs is usually needed to maintain sinus rhythm.91 Although the ideal of pharmacologic therapy would be to prevent all recurrences of AF, this is unrealistic for many patients. Rather, marked reduction of the frequency, duration, and symptoms of AF may be a very acceptable clinical goal. In addition, the use of pharmacologic agents resulting in an apparent clinical resolution of AF does not change the indication for anticoagulation.
The choice of pharmacologic agent is largely determined by the potential side effects of a given drug in an individual patient. The first drug chosen is usually associated with a low risk of serious side effects for that patient. For most patients, a specific etiologic factor can not be identified for the initiation of AF. Conversely, if such an inciting factor is uncovered, therapeutic efforts should be targeted to eliminating it. Examples may be β-blockers for exercise-induced AF or avoidance of alcohol in selected individuals. Many agents have effectiveness to maintain sinus rhythm in patients with AF.3,92-96 Because class IC drugs may suppress AF but promote atrial flutter, it is combined with a β-blocker or calcium channel blocker to decrease the risk of atrial flutter with 1:1 ventricular response, a potentially life-threatening situation. Monitoring the QRS duration and PR interval are important during class IC therapy. If the QRS duration increases to 150% of the baseline value, reduce the dosage or discontinue the drug. Sotalol, dofetilide, and amiodarone prolong ventricular refractoriness and the QT interval. Monitoring the QT interval during initiation of therapy is important. If possible, avoid corrected QT intervals of >500 to 520 ms with sotalol and dofetilide, but longer QT intervals may occur without a risk of proarrhythmia in patients receiving amiodarone. Periodic ECGs should be obtained on an outpatient basis in patients receiving antiarrhythmic drugs, and efforts to avoid hypokalemia are important.
Dronedarone is a new antiarrhythmic drug recently approved for the treatment of patients with AF and atrial flutter. It is a noniodinated benzofuran derivative of amiodarone.97 Like amiodarone, it blocks multiple channels, including sodium, potassium, and calcium and has noncompetitive antiadrenergic effects. Compared with placebo in two randomized controlled trials, dronedarone prolonged the median times to recurrence of arrhythmia.98 In a multicenter randomized controlled trial of more than 4600 patients, dronedarone reduced the incidence of hospitalization due to cardiovascular events or death in patients who had atrial fibrillation.99 Based on the study of recently hospitalized patients with heart failure who were randomized to dronedarone or placebo, and in which those treated with dronedarone had a worse mortality,100 dronedarone is contraindicated in patients with New York Heart Association (NYHA) class IV heart failure or NYHA class II to III heart failure, with recent decompensation requiring hospitalization or referral to a specialized heart failure clinic.
There are not many prospective data on the safety of initiating drugs in the outpatient setting, but some general rules are useful. Flecainide and propafenone can be initiated in patients without heart disease who are in sinus rhythm, and safety is maximized if AV nodal blockers are given first. Amiodarone can be used in patients with or without sinus rhythm, but frequent ECG monitoring, for example, by using event recorders, is recommended to identify any potential bradycardia or tachycardia proarrhythmia. Sotalol may be administered to patients in sinus rhythm with minimal or no heart disease and normal QT interval and electrolyte status; the lowest dose should be used and an ECG should be obtained within days of starting sotalol to determine the QT interval. The same process should occur for any dose increase. Dofetilide must be started in hospital, and dronedarone is typically started out of hospital.
Antiarrhythmic drugs are selected on a safety-first basis (Fig. 40–11). The ACC/AHA/ ESC guidelines3 suggest for patients with no or minimal heart disease to start with flecainide, propafenone, or sotalol, agents with minimal noncardiac toxicity. The second-line therapy is either amiodarone/dofetilide or catheter ablation. Patients with hypertension who do not have substantial LV hypertrophy have a similar treatment algorithm, but those with substantial LV hypertrophy are considered at increased proarrhythmia risk with most drugs other than amiodarone, which becomes first-line therapy here. Catheter ablation is second-line treatment. Safety of drugs in coronary artery disease has been demonstrated for dofetilide/sotalol (first line) and amiodarone (second line), and catheter ablation is also second-line treatment. For patients with heart failure, first-line treatment can be either amiodarone or dofetilide, but the authors prefer amiodarone in most circumstances, and ablation is second-line therapy. The agents quinidine, procainamide, and disopyramide are currently considered as drugs of last resort. As dronedarone has only recently been approved for marketing, optimal patient selection for its use is still evolving, but it appears to be a safe drug to use as first-line therapy in all patient subgroups, except those who have heart failure contraindications. At present, we think it should be used with great caution, if at all, in patients with NYHA class III heart failure.
Figure 40–11
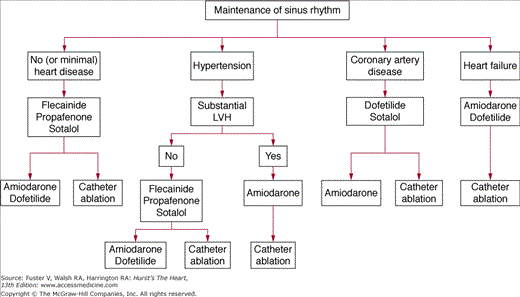
Proposed strategy for use of antiarrhythmic drugs to maintain sinus rhythm in patients with atrial fibrillation. LVH, left ventricular hypertrophy. Modified with permission from Fuster et al.3
Several drugs used to treat other medical conditions have shown promise as adjuvant therapy in patients with AF. Drugs that modulate the rennin-angiotensin-aldosterone system, for example, angiotensin-converting enzyme inhibitors and angiotensin-receptor blockers, may decrease the incidence of AF.45,101-107
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


