Atherosclerotic Biology and Epidemiology of Disease
James H.F. Rudd
John R. Davies
Peter L. Weissberg
Epidemiology of Cardiovascular Disease
Atherosclerosis, with its complications, is the leading cause of mortality and morbidity in the developed world. In the United States, a snapshot of the population reveals that 60 million adults currently suffer from atherosclerotic cardiovascular disease, which accounts for 42% of all deaths annually, at a cost to the nation of $128 billion. Fortunately, despite this catastrophic burden of disease, much evidence has emerged over the last decade suggesting that the progression of atherosclerosis can be slowed or even reversed in many people with appropriate lifestyle and drug interventions.
The origin of the current epidemic of cardiovascular disease can be traced back to the time of industrialization in the 1700s. The three factors largely responsible for this were an increase in the use of tobacco products, reduced physical activity, and the adoption of a diet high in fat, calories, and cholesterol. This rising tide of cardiovascular disease continued into the twentieth century, but began to recede when data from the Framingham study identified a number of modifiable risk factors for cardiovascular disease, including cigarette smoking, hypertension, and hypercholesterolemia (1).
The number of deaths per 100,000 attributable to cardiovascular disease peaked in the Western world in 1964 to 1965, since which time there has been a gradual decline in death rates (Fig. 1.1) (2). The age-adjusted coronary heart disease (CHD) mortality in the United States dropped by more than 40% and cerebrovascular disease mortality by more than 50%, with the greatest reductions being seen among whites and men. This reduction has occurred despite a quadrupling of the proportion of the population older than 65 years of age and has been due to a number of factors, particularly major health promotion campaigns aimed at reducing the prevalence of Framingham risk factors. Indeed, there has been a substantial change in the prevalence of population cardiovascular risk factors over the last 30 years (Table 1.1). The war is not won, however, and the decline in the death rate from cardiovascular disease slowed in the 1990s (Fig. 1.2). This is likely owing to a large increase in the prevalence of both obesity and type 2 diabetes mellitus, as well as a resurgence of cigarette smoking in some sectors of society (3). Female death rates from cardiovascular disease overtook male death rates in 1984 and have shown a smaller decline over the last 30 years (4). The consequences of atherosclerosis are also beginning to be felt in less well-developed regions of the globe (5), with death from atherosclerotic cardiovascular disease set to replace infection as the leading cause of death in the Third World in the near future. This phenomenon is further illustrated by the increase in CHD mortality in countries of Eastern and Central Europe (most notably countries of the former Soviet Union). For example, in the Ukraine the age standardized death rate in the year 2000 was just over 800 per 100,000 people representing an increase of over 60% when compared to 1990 (6).
A further note of caution should also be struck. Western countries are experiencing a dramatic increase in the prevalence of heart failure. In the United States, almost 5 million people carry a diagnosis of heart failure (7), thus singling it out as an emerging epidemic (8). However, the determinants of this epidemic have yet to be fully elucidated, with some epidemiologic studies pointing toward hypertension as the driving factor (9) and others suggesting CHD as the predominant cause (10).
Biology of Atherosclerosis
Traditionally, atherosclerosis has been viewed as a degenerative disease, affecting predominantly older people, slowly progressing over many years, and eventually leading to symptoms through mechanical effects of blood flow. The perceived insidious and relentless nature of its development has meant that
a somewhat pessimistic view of the potential to modify its progression by medical therapy has held sway. There has been little emphasis on the diagnosis and treatment of high-risk asymptomatic patients. Disease management has instead been dominated by interventional revascularization approaches, targeting the largest and most visible or symptomatic lesions with coronary angioplasty or bypass surgery.
a somewhat pessimistic view of the potential to modify its progression by medical therapy has held sway. There has been little emphasis on the diagnosis and treatment of high-risk asymptomatic patients. Disease management has instead been dominated by interventional revascularization approaches, targeting the largest and most visible or symptomatic lesions with coronary angioplasty or bypass surgery.
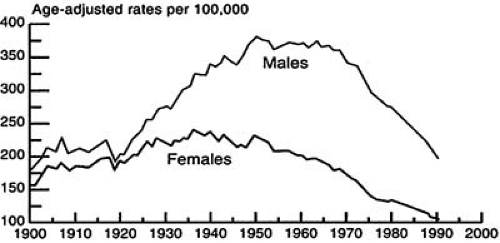 FIGURE 1.1. Trends in death rates for heart diseases: United States, 1900–1991. (Source: Feinleib M. Trends in heart disease in the United States [review]. Am J Med Sci 1995;310[Suppl 1]:S8–S14 , with permission.) |
Recently, for several reasons, this defeatist view of the pathogenesis and progression of atherosclerosis has begun to change. First, careful descriptive studies of the underlying pathology of atherosclerosis have revealed that atherosclerotic plaques differ in their cellular composition and that the cell types predominating in the plaque can determine the risk of fatal clinical events. A high degree of plaque inflammation is particularly dangerous. Second, recent epidemiologic work has identified many new potentially modifiable risk factors for atherosclerosis, above and beyond those highlighted as a result of the Framingham study (11). The third and most important reason is because several large-scale clinical trials have reported that drugs—in particular, the HMG-CoA reductase inhibitors (statins)—are able to reduce the number of clinical events in patients with established atherosclerosis and do so without necessarily affecting the size of atherosclerotic plaques. These three strands of evidence have shown that, rather than being an irreversibly progressive disease, atherosclerosis is a dynamic, inflammatory process that may be amenable to medical therapy. Understanding the cellular and molecular interactions that determine the development and progression of atherosclerosis brings with it opportunities to develop novel therapeutic agents targeting key molecular and cellular interactions in its etiology. In addition, the recognition that the clinical consequences of atherosclerosis depend almost entirely on plaque composition argues for a new approach to diagnosis, with less emphasis placed on the degree of lumen narrowing and more interest in the cellular composition of the plaque.
TABLE 1.1 Temporal Changes in Coronary Risk Factors | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Normal Artery
The healthy artery consists of three histologically distinct layers. Innermost and surrounding the lumen is the tunica intima, which comprises a single layer of endothelial cells in close proximity to the internal elastic lamina. The tunica media surrounds the internal elastic lamina, and its composition varies depending on the type of artery. The tunica media of the smallest arterial vessels, arterioles, comprises a single layer of vascular smooth muscle cells (VSMCs). Small arteries have a similar structure but with a thicker layer of medial VSMCs. Arterioles and small arteries are termed resistance vessels because they contribute vascular resistance and, hence, directly affect blood pressure. At the opposite end of the spectrum are large elastic or conduit arteries, named for the high proportion of elastin in the tunica media. The tunica media of all arteries is contained within a connective tissue layer that contains blood vessels and nerves and that is known as the tunica adventitia. In normal arteries, the vessel lumen diameter can be altered by contraction and relaxation of the medial VSMCs in response to a variety of systemic and locally released signals.
Atherosclerotic Vessel
Atherosclerosis is primarily a disease affecting the intimal layer of elastic arteries. For reasons that remain largely unknown, some arterial beds appear more prone than others. Coronary, carotid, cerebral, and renal arteries and the aorta are most often involved. The arteries supplying the lower limbs are also vulnerable to disease. Interestingly, the internal mammary artery is almost always spared, making it an invaluable vessel for coronary bypass surgery.
Atherosclerotic lesions develop over many years and pass through several overlapping stages. Histologically, the earliest lesion is a subendothelial accumulation of lipid-laden macrophage foam cells and associated T lymphocytes known as a fatty streak. Fatty streaks are asymptomatic and nonstenotic. Postmortem examinations have shown that they are present in the aorta at the end of the first decade of life, are present in the coronary arteries by the second, and begin to appear in the cerebral circulation by the third decade. With time, the lesion progresses and the core of the early plaque becomes necrotic, containing cellular debris, crystalline cholesterol, and inflammatory cells, particularly macrophage foam cells. This necrotic core becomes bounded on its luminal aspect by an endothelialized fibrous cap, consisting of VSMCs embedded in an extensive collagenous extracellular matrix. Inflammatory cells are also present in the fibrous cap, concentrated particularly in the “shoulder” regions, where T cells, mast cells, and especially macrophages have a tendency to accumulate. Advanced lesions may become increasingly complex, showing evidence of calcification, ulceration, new microvessel formation, and rupture or erosion (12). Microvessels within the plaque may play important roles in the formation of macrophage-rich vulnerable atheroma by providing an extended surface area of activated endothelial cells to hasten recruitment of further inflammatory cells as well as by promotion of intraplaque hemorrhage (13).
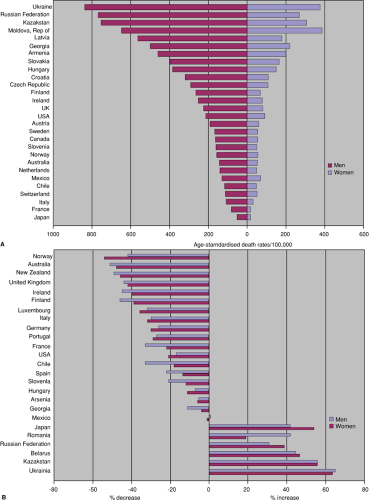 FIGURE 1.2. A. Death rates from CHD, men and women aged 35–74, 2000, selected countries. B. Changes in death rates from CHD, men and women aged 35–74, between 1990 and 2000, selected countries. |
Thus, the composition of atherosclerotic plaques is variable, dynamic and complex, and it is the interaction between the various cell types within a plaque that determines the progression, complications, and outcome of the disease.
Cellular Roles in Atherogenesis
Endothelial Cells
The endothelium plays a central role in maintaining vascular health by virtue of its vital anti-inflammatory and anticoagulant properties. Many of these characteristics are mediated by the nitric oxide (NO) molecule. This molecule was discovered in the 1980s, having been isolated from lipopolysaccharide-primed macrophages (14). NO is synthesized by endothelial cells under the control of the enzyme endothelial NO synthase (NOS) and has a number of anti-atherogenic properties. First, it acts as a powerful inhibitor of platelet aggregation on endothelial cells. Second, it can reduce inflammatory cell recruitment into the intima by abrogating the expression of genes involved in this process, such as those encoding intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), P-selectin, and monocyte chemoattractant protein-1 (MCP-1) (15,16,17). There is some evidence that NO may also reduce lipid entry into the arterial intima (18). NO is also a potent anti-inflammatory molecule and, depending on concentration, may be a scavenger or a producer of potentially destructive oxygen free radicals, such as peroxynitrite (19,20,21). The earliest detectable manifestation of atherosclerosis is a decrease in the bioavailability of NO in response to pharmacologic or hemodynamic stimuli (22). This may occur for two reasons. Either there may be decreased manufacture of NO because of endothelial cell dysfunction, or increased NO breakdown may take place. There is evidence that both mechanisms may be important in different situations (23). Many atherosclerosis risk factors can lead to impaired endothelial function and reduced NO bioavailability. For example, hyperlipidemic patients have reduced NO-dependent vasodilatation, which is reversed when patients are treated with lipid-lowering medication (24). Diabetics also have impaired endothelial function, occurring primarily as a result of impaired NO production. There is, however, some evidence to suggest that increased oxidative stress leading to enhanced NO breakdown may also be a factor in early endothelial dysfunction (25). Similarly, other risk factors for atherosclerosis, such as hypertension and cigarette smoking, are associated with reduced NO bioavailability (26,27). In cigarette smokers, endothelial impairment is thought to be caused by enhanced NO degradation by oxygen-derived free-radical agents such as the superoxide ion. There are also other consequences of an increased reactivity between NO and superoxide species. The product of their interaction, ONOO– (peroxynitrite), is a powerful oxidizing agent and can reach high concentrations in atherosclerotic lesions. This may result in cellular oxidative injury.
Another consequence of endothelial cell dysfunction that occurs in early atherosclerosis is the expression of surface-bound selectins and adhesion molecules, including P-selectin, ICAM-1, and VCAM-1. These molecules attract and capture circulating inflammatory cells and facilitate their migration into the subendothelial space (22). Normal endothelial cells do not express these molecules, but their appearance may be induced by abnormal arterial shear stress, subendothelial oxidized lipid, and, in diabetic patients, the presence of advanced glycosylation products in the arterial wall. The importance of selectins and adhesion molecules in the development of atherosclerosis is demonstrated in experiments using mice, which lack their expression. These animals develop smaller lesions with a lower lipid content and fewer inflammatory cells than control mice when fed a lipid-rich diet (28). Animal models have reinforced the importance of inflammatory cell recruitment to the pathogenesis of atherosclerosis, but because inflammatory cells are never seen in the intima in the absence of lipid, the results suggest that subendothelial lipid accumulation is also necessary for the development of atherosclerosis.
The tendency for atherosclerosis to occur preferentially in particular sites may be explained by subtle variations in endothelial function. This is probably caused by variations in local blood flow patterns, especially conditions of low flow, which can influence expression of a number of endothelial cell genes, including those encoding ICAM-1 and endothelial NOS (29,30). In addition to flow speed, flow type can have a direct effect on cell morphology. In areas of laminar flow (atheroprotective flow), endothelial cells tend to have an ellipsoid shape, contrasting with the situation found at vessel branch points and curves, where turbulent flow (atherogenic flow) induces a conformational change toward polygonal-shaped cells (31). Such cells have an increased permeability to low-density lipoprotein (LDL) cholesterol and may promote lesion formation (32).
These data are consistent with the idea that the primary event in atherogenesis is endothelial dysfunction. The endothelium can be damaged by a variety of means, leading to dysfunction and, by unknown mechanisms, subsequent subendothelial lipid accumulation. In this situation, the normal homeostatic features of the endothelium break down; it becomes more adhesive to inflammatory cells and platelets, it loses its anticoagulant properties, and there is reduced bioavailability of NO. Importantly, endothelial function is improved by drugs that have been shown to substantially reduce death from vascular disease, including statins and angiotensin-converting enzyme inhibitors (33,34).
Inflammatory Cells
LDL from the circulation is able to diffuse passively through the tight junctions that bind neighboring endothelial cells. The rate of passive diffusion is increased when circulating levels of LDL are elevated. In addition, other lipid fractions may be important in atherosclerosis. Lipoprotein(a) has the same basic molecular structure as LDL, with an additional apolipoprotein(a) element attached by a disulfide bridge. It has been shown to be highly atherogenic (35), accumulate in the arterial wall in a manner similar to LDL (36), impair vessel fibrinolysis (37), and stimulate smooth muscle cell proliferation (38). The accumulation of subendothelial lipids, particularly when at least partly oxidized, is thought to stimulate the local inflammatory reaction that initiates and maintains activation of overlying endothelial cells. The activated cells express a variety of selectins and adhesion molecules and also produce a number of chemokines—in particular, MCP-1, whose expression is upregulated by the presence of oxidized LDL in the subendothelial space (39). Interestingly, the protective effect of high-density lipoprotein (HDL) against atherosclerotic vascular disease may be partly explained by its ability to block endothelial cell expression of adhesion molecules (40,41). Chemokines are proinflammatory cytokines responsible for chemoattraction, migration, and subsequent activation of leukocytes. Mice lacking the MCP-1 gene develop smaller atherosclerotic lesions than normal animals (42). The first stage of inflammatory cell recruitment to the intima is the initiation of “rolling” of monocytes and T cells along the endothelial cell layer. This phenomenon is mediated
by the selectin molecules, which selectively bind ligands found on these inflammatory cells. The subsequent firm adhesion to and migration of leukocytes through the endothelial cell layer depends on the endothelial expression of adhesion molecules such as ICAM-1 and VCAM-1 and their binding to appropriate receptors on inflammatory cells. Once present in the intima, monocytes differentiate into macrophages under the influence of chemokines such as macrophage colony-stimulating factor. Such molecules also stimulate the expression of the scavenger receptors that allow macrophages to ingest oxidized lipids and to develop into macrophage foam cells, the predominant cell in an early atherosclerotic lesion. The formation of scavenger receptors is also regulated by peroxisome proliferator-activated receptor-γ (PPAR-γ a nuclear transcription factor expressed at high levels in foam cells) (43). PPAR-γ agonists (glitazones), which are used to treat patients with type 2 diabetes, have been shown to have many anti-atherogenic effects, including increasing production of NO (44), decreased endothelial inflammatory cell recruitment and reduced vascular endothelial growth factor (VEGF) expression (45). Also, PPAR-γ agonists can reduce the lipid content of plaques by enhancing reverse cholesterol transport from plaque to liver. Positive results with these drugs in patients with type 2 diabetes are emerging. As well as reducing matrix metalloproteinase (MMP) 9 levels, glitazones also significantly ameliorated C-reactive protein (CRP) and CD40 ligand levels, as well as causing direct plaque regression in a rabbit atheroma model (46). Clearly their use in large clinical trials in patients without diabetes as anti-atheroma drugs is awaited with interest.
by the selectin molecules, which selectively bind ligands found on these inflammatory cells. The subsequent firm adhesion to and migration of leukocytes through the endothelial cell layer depends on the endothelial expression of adhesion molecules such as ICAM-1 and VCAM-1 and their binding to appropriate receptors on inflammatory cells. Once present in the intima, monocytes differentiate into macrophages under the influence of chemokines such as macrophage colony-stimulating factor. Such molecules also stimulate the expression of the scavenger receptors that allow macrophages to ingest oxidized lipids and to develop into macrophage foam cells, the predominant cell in an early atherosclerotic lesion. The formation of scavenger receptors is also regulated by peroxisome proliferator-activated receptor-γ (PPAR-γ a nuclear transcription factor expressed at high levels in foam cells) (43). PPAR-γ agonists (glitazones), which are used to treat patients with type 2 diabetes, have been shown to have many anti-atherogenic effects, including increasing production of NO (44), decreased endothelial inflammatory cell recruitment and reduced vascular endothelial growth factor (VEGF) expression (45). Also, PPAR-γ agonists can reduce the lipid content of plaques by enhancing reverse cholesterol transport from plaque to liver. Positive results with these drugs in patients with type 2 diabetes are emerging. As well as reducing matrix metalloproteinase (MMP) 9 levels, glitazones also significantly ameliorated C-reactive protein (CRP) and CD40 ligand levels, as well as causing direct plaque regression in a rabbit atheroma model (46). Clearly their use in large clinical trials in patients without diabetes as anti-atheroma drugs is awaited with interest.
In early atherosclerosis at least, the macrophage can be thought of as performing a predominantly beneficial role as a “neutralizer” of potentially harmful oxidized lipid components in the vessel wall. However, macrophage foam cells also synthesize a variety of proinflammatory cytokines and growth factors that contribute both beneficially and detrimentally to the evolution of the plaque. Some of these factors are chemoattractant (osteopontin) and growth-enhancing (platelet-derived growth factor) for VSMCs (12,47). Under the influence of these cytokines, VSMCs migrate from the media to the intima, where they adopt a synthetic phenotype, well-suited to matrix production and protective fibrous cap formation.
However, activated macrophages have a high rate of apoptosis. Once dead, they release their lipid content, which becomes part of the core of the plaque, thereby contributing to its enlargement. The apoptotic cells also contain high concentrations of tissue factor, which may invoke thrombosis if exposed to circulating platelets (48). Interestingly, the selective glycoprotein 2b3a receptor antagonist abciximab has been shown to have an effect on the levels of tissue factor found in monocytes. In an in vitro study by Steiner (49), the drug attenuated both the amount of tissue factor and its RNA levels. As tissue factor is a potent instigator of the clotting cascade, this role may explain part of the protective effect of abciximab on the microcirculation of patients with acute coronary syndromes (50).
It is now generally recognized that the pathologic progression and consequences of atherosclerotic lesions are determined by dynamic interactions between inflammatory cells recruited in response to subendothelial lipid accumulation, and the local reparative “wound healing” response of surrounding VSMCs.
Vascular Smooth Muscle Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


