Chapter 49 Asymptomatic Electrocardiogram Abnormalities
Introduction
Asymptomatic Wolff-Parkinson-White Pattern
Case 1
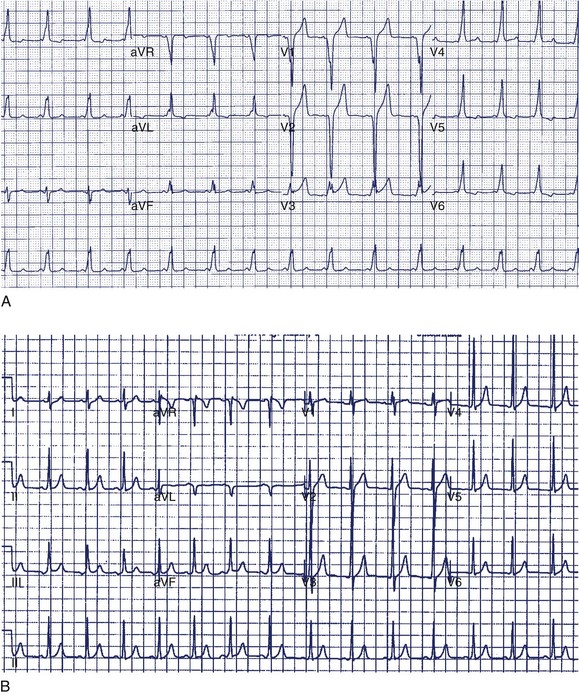
A 14-year-old boy was seen in the emergency department because of noncardiac chest pain following chest trauma during a basketball game. The electrocardiogram (ECG) seen in Figure 49-1 was recorded. He had never been aware of palpitations or syncope. Electrocardiographic pre-excitation was noted.
The most appropriate next step would be to:
The ECG findings now known as ventricular pre-excitation (δ-wave and short P-R interval) were first described in 1930.1 Initial interest was in the arrhythmias associated with the ECG pattern, although it was later observed that many individuals with identical ECG patterns remained asymptomatic and had a benign course. Radiofrequency (RF) ablation of accessory pathways has become the preferred treatment option for patients with Wolff-Parkinson-White (WPW) syndrome because it is curative and has a low rate of associated complications. The question that remains is whether this treatment should be extended as a prophylactic measure to asymptomatic individuals with the WPW pattern on ECG to prevent the small risk of sudden cardiac death (SCD).
Several studies have provided an estimate of the prevalence of the WPW pattern. It is estimated that 0.1% to 0.15% of the population will have ECG manifestations of WPW, of which approximately 50% will never have had symptoms.2 New cases arise at a rate of approximately 0.004% per year.2 However, the true incidence is likely higher, given that the WPW pattern is frequently intermittent and may be missed on ECG and that many individuals never receive an ECG in their lifetime.
Although estimates vary, the incidence of SCD among patients with WPW appears to be at most 0.1% per year.2,3 Munger et al did not report any SCDs in their large (113 patients) retrospective review. No deaths occurred among 293 patients studied in a recent prospective study that examined the usefulness of the EPS and ablation in asymptomatic individuals.4 Autopsy studies delving into the issue are generally limited by the difficulty in finding accessory pathways on routine tissue sections, but most cases of unexpected SCD are related to coronary or other unrecognized structural heart diseases. Nonetheless, SCD in an asymptomatic young individual with WPW pattern is a tragic event that cannot be ignored.
The most common cause of SCD in patients with WPW is ventricular fibrillation (VF) triggered by atrial fibrillation (AF). In these cases, AF is associated with a rapid ventricular response because of the presence of at least one accessory pathway with a short anterograde refractory period. It follows that only those patients who have accessory pathways capable of mediating a rapid ventricular rate are at risk for developing SCD. It has been found that VF rarely occurs when the shortest R-R (SRR) interval during induced AF is greater than 250 ms.5 AF is usually preceded by AV re-entrant tachycardia in these patients, presumably because of the electrophysiological, hemodynamic, and metabolic consequences of supraventricular tachycardia (SVT). Hence, the failure to induce tachycardia would independently predict a good prognosis.6 Interestingly, the presence of multiple accessory pathways in a given individual increases the risk of SCD by an odds ratio of 3.1, presumably because of increased ventricular desynchronization related to multiple pre-excitation wavefronts.7
Because of the rarity of the outcome in question (<1/1000 patients/year) it is evident that predicting outcomes with any test is problematic. As one would expect from such a low incidence of SCD, the negative predictive value of the EPS is excellent. Klein et al have categorized the risk of SCD on the basis of the SRR interval during spontaneous or induced AF as follows8:
It must be emphasized that “definite” risk in this stratification is not equivalent to “high” risk. The use of the SRR less than 250 ms as a marker of risk in clinical practice is not as helpful as may be believed; 17% of asymptomatic individuals with WPW will have SRRs in this range, a far greater proportion than that expected to have an event.8 Using an SRR of less than 250 ms, sensitivity of 77.8%, specificity of 48.3%, and predictive accuracy of 18.9% can be obtained.9 Combining the findings of multiple accessory pathways and SRR less than 250 ms lowered the sensitivity (from 88% to 29%) but increased the specificity (from 36% to 92%) and the positive predictive value (from 9% to 22%).7 Pappone et al reported that the combination of a short pathway effective refractory period (ERP) (<250 ms) and inducible tachycardia had sensitivity of 93.7%, specificity of 67.6%, and positive and negative predictive values of 46.9% and 97.3%, respectively, for subsequent arrhythmia.6 Isoproterenol, which is often used in an attempt to induce tachycardia, AF, or both, will cause an even greater percentage of individuals to have an SRR less than 250 ms. Therefore the already poor positive predictive value of an SRR less than 250 ms can be expected to worsen. Despite its limitations, the EPS remains the gold standard for the determination of risk of SCD in asymptomatic patients with WPW.
Noninvasive methods for evaluating the risk for SCD have been described: Holter monitoring to determine whether intermittent pre-excitation is present (therefore making it extremely unlikely for the SRR of the pathway to be <250 ms), sudden loss of pre-excitation on an exercise test, with infusion of ajmaline or procainamide (suggesting a long refractory period for the pathway), and trans-esophageal pacing.10–12 With the exception of trans-esophageal pacing, all of these methods rely on assessments of accessory pathway conduction in sinus rhythm and are imperfect substitutes for a test that, in itself, has limitations. Trans-esophageal pacing has its proponents, but it provides less information than catheter EPS and arguably involves similar risk and discomfort.13
What, then, is the best approach for the management of the asymptomatic patient, given that SCD as a first manifestation of the WPW syndrome is a very rare occurrence? Since serious complications from RF ablation in experienced centers occur infrequently (~1%), it becomes difficult to make a categorical recommendation for or against ablation. Clearly, studying all patients with asymptomatic WPW and ablating those with an SRR less than 250 ms will result in increased morbidity and mortality among patients who may never develop any symptoms. However, an approach that involves not studying any asymptomatic individuals may result in some preventable deaths. Many physicians may prefer to accept a small procedural risk with a finite risk interval over a longer term risk of potentially lethal arrhythmias.14,15
Finally, two other factors may influence the discussion:
Discussion of Case 1
In this case, the δ-wave was prominent and could be easily monitored during a treadmill test. This is the least-invasive and most appropriate next step. If loss of pre-excitation occurs with exercise, this classifies the accessory pathway as benign and eliminates concerns of SCD. An EPS is probably premature at this point but may be required if the treadmill test is not prognostically helpful. Treadmill testing is particularly problematic when minimal pre-excitation occurs (see Figure 49-1, B). In such cases, an EPS may be the only method of risk evaluation. An echocardiogram is not useful under the circumstances because most patients with WPW have structurally normal hearts.
Repolarization Abnormalities
Many primary repolarization changes on the 12-lead ECG are benign or normal variants such as early repolarization. Others may result from the presence of structural heart disease (e.g., myocardial infarction [MI], dilated cardiomyopathy, and hypertrophic cardiomyopathy) or the consequence of pharmacologic agents (e.g., antiarrhythmic drugs). The congenital long QT syndrome (LQTS), short QT syndrome (SQTS), and Brugada syndrome are relatively uncommon diseases associated with potentially life-threatening ventricular arrhythmias. In general, diagnoses of these conditions are complicated because of the significant overlap in the ECG manifestations of normal and disease conditions. The management of these conditions continues to evolve. The following sections discuss the typical ECG changes and clinical features that accompany the most common pathologic repolarization abnormalities. Readers are also directed to Chapters 62 to 64 for additional information.
Long QT Syndrome
Case 2
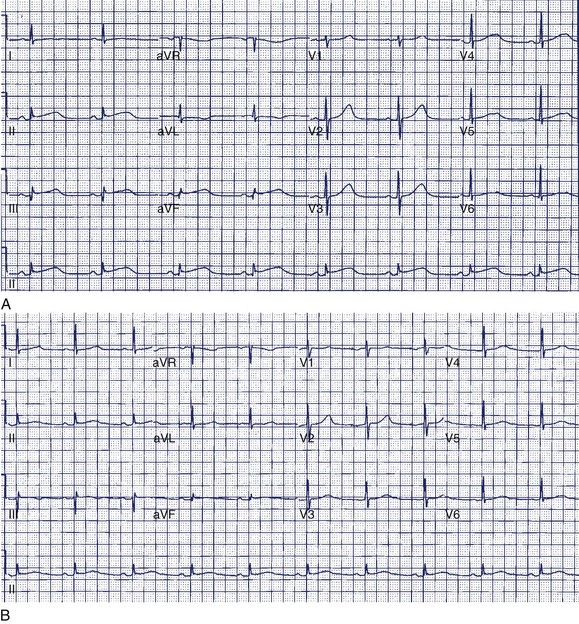
A 36-year-old accountant was referred for assessment after the unexpected death of his 24-year-old brother who was previously well. The postmortem examination revealed no obvious cause of death. A 27-year-old sister is alive and well. His mother, who was known to have epilepsy, had died at age 46 in a motor vehicle accident. The patient’s ECG shows a very long Q-T interval (QT = 600 ms, QTc = 545 ms) (Figure 49-2, A). He was not taking any medication and jogged regularly. An ECG done 2 years previously for an insurance physical was reported as normal (Figure 49-2, B).
The most appropriate course of action at this time would be to:
The congenital LQTS is a rare disorder (incidence 1 : 3000 to 1 : 10 000) characterized by prolongation of the Q-T interval, recurrent syncope, and SCD.16 Two major clinical variants were originally described: (1) Romano-Ward syndrome (autosomal dominant inheritance) and (2) Jervell Lange-Nielson syndrome (autosomal recessive inheritance), in which patients also have congenital deafness. Over the past 10 years, great advances have been made in understanding the genetic and cellular bases for LQTS. At least 10 distinct genotypic variants of LQTS encompassing hundreds of mutations have now been identified (Table 49-1), and a genetic diagnosis is now obtainable in 50% to 60% of patients.17 Three genotypes (LQT1, LQT2, and LQT3) account for more than 90% of cases.18–20 The principal abnormality in LQTS is prolongation of the action potential duration caused by a reduction in the outward potassium current (LQT1 and LQT2) or, less commonly, by a persistent inward sodium current during the plateau phase (LQT3). Action potential prolongation provokes the development of early afterdepolarizations, which may produce a triggered action potential resulting in a premature beat.21 Such impulses can initiate a polymorphic VT, known as torsades de pointes, which underlies the clinical symptoms of palpitations, syncope, or SCD caused by VF.
The classic description of events in LQTS involves exercise-induced or emotion-induced syncope or cardiac arrest. Symptoms often begin in adolescence, though they may begin earlier in patients with LQT1.20 It is common for patients to be investigated and misdiagnosed as having a seizure disorder. Clinical events in LQTS are often seen in the context of heart rate acceleration precipitated by exercise, emotion, or sudden arousal.20,22,23 Patients with LQT1 are particularly prone to events during hyperadrenergic states, with swimming identified as a specific trigger in such patients.20,23 The history of arousal to an auditory stimulus such as a sudden loud noise (classically, a telephone call at night or an alarm clock going off) is strongly predictive of LQT2.20,22 Events that occur during rest or sleep are more suggestive of LQT3.20 Many of these circumstances represent the setting of sudden or marked changes in adrenergic tone and heart rate that may influence repolarization and subsequent vulnerability to arrhythmia.
The hallmark of LQTS is prolongation of the QTc (>440 ms in men and >460 ms in women), although considerable overlap occurs with QTc in normal populations. The QTc of genotype-positive patients overlaps with normal in up to one third because of variable penetrance.24–26 Also, 10% to 15% of QTc in normal adults are greater than 440 ms.27 Since repolarization is affected by factors such as sympathetic outflow, electrolyte balance, and pharmacologic agents, it is not surprising that considerable temporal variation may be present in QTc and that increased sampling improves diagnostic accuracy. In the absence of obvious reversible causes of QTc prolongation (Table 49-2), a diagnosis of LQTS can be made on the basis of ECG features and clinical presentation.28 The mean QTc does not differ among the LQT1, LQT2, and LQT3 types but is significantly longer in Jervell Lange-Neilson syndrome.20 In addition to QTc prolongation, qualitative abnormalities may also be found in LQTS, including ST-T wave changes, U waves, T-wave alternans, increased QT dispersion, and sinus bradycardia.21 Although not highly specific, characteristic ST-T wave morphologies may allow the prediction of LQT type: broad-based T waves typify LQT1; low amplitude, notched T waves occur in LQT2; and long isoelectric ST segment with late-onset T wave occur in LQT3.26
Table 49-2 Secondary Causes of QT Prolongation
| FACTOR | MECHANISM |
|---|---|
| Bradycardia | ↑ APD |
| ↑ APD prolongation with class III agents | |
| Drugs* | Mainly IKr blockade |
| Electrolyte disorders (hypokalemia, hypomagnesemia, hypocalcemia) | Hypokalemia ↓ IKr and ↑ IKr sensitivity to pharmacologic blockers |
| Left ventricular hypertrophy/failure | ↓ K+ currents (Ito, IKr, IKs) |
| Changes to ICaL and intracellular calcium (Ca2+) | |
| Miscellaneous (e.g., anorexia, cerebrovascular disease, HIV infection, hypothyroidism, hypothermia, ionic contrast media) |
APD, Action potential duration.
* The major classes of drugs are antiarrhythmic drugs, antihistamines, macrolide antibiotics, antipsychotics, and antidepressants. An updated online list of specific drugs that prolong the Q-T interval is available at www.qtdrugs.org or www.torsades.org.
Modified from Walker BD, Krahn AD, Klein GJ, et al: Congenital and acquired long QT syndromes, Can J Cardiol 19(1):76–87, 2003.
It has been estimated that LQTS causes 3000 to 4000 SCDs per year in children and young adults in the United States.29 Long-term registry data suggest that annually the risk of syncope is 5% and the risk of LQTS-related death before the age of 50 years is approximately 1% in symptomatic patients, with the risk being significantly lower in asymptomatic patients.26,30 The most important predictor of risk is QTc duration, although age, gender, and genotype can be confounders. Using data from an international registry, three risk groups relating to the probability of experiencing a first cardiac event before the age of 40 years have been identified31:
Although a family history of cardiac events would intuitively appear to be prognostically important, this has not been borne out in clinical studies.32,33
β-Adrenoreceptor blockade is the mainstay of therapy (aiming for a reduction in peak exercise heart rate of >20%), and it is recommended for most patients with LQTS.34 Survival can be dramatically improved with aggressive treatment with β-blockers, left cervical sympathectomy, pacemakers, and an implantable cardioverter defibrillator (ICD).17,34 Lifestyle modifications, family screening, and genetic testing and counseling are also important considerations. In particular, genotyping may occasionally assist with diagnosis, family screening, and management.35 For example, β-blockers may be less effective in LQT3, and genotype-specific therapies such as potassium replacement for LQT2 and mexiletine for LQT3 are being evaluated.36,37
Discussion Case 2
The patient in question clearly has marked QTc prolongation in the absence of medication. The family history is a cause for concern and suggests that his brother and mother (LQTS is frequently misdiagnosed as epilepsy) both had LQTS. This man previously had a normal ECG, and it is not unusual for such patients to have variability of the Q-T interval at different times. In more borderline cases, scoring systems incorporating ECG, clinical features, and family history may be helpful.28 In this case, the best answer would be to recommend treatment with β-blocker. An argument can be made for an ICD in such a patient, although ICDs are usually reserved for high-risk patients such as those with recurrent symptoms who are on β-blockers or survivors of cardiac arrest (class IIa recommendation). Treadmill testing will not influence the decision to treat, although exercise may be diagnostically useful, especially if arrhythmias are observed. The EPS is generally of no value in LQTS. Where available, genetic counseling and family screening should also be offered.
Brugada Syndrome
Case 3
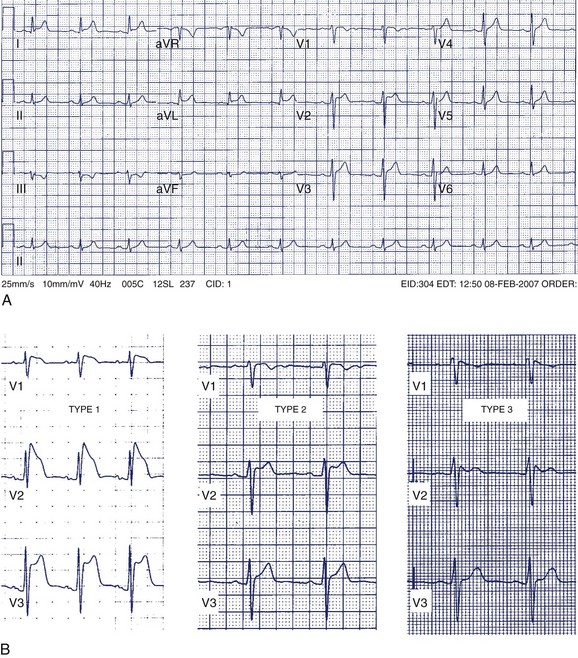
A 42-year-old man was referred for assessment of symptoms of atypical chest pain and ECG abnormalities (Figure 49-3). The patient was otherwise well. His father died suddenly in his mid-40s but no further details about his death are available.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


