Chapter 57 Arrhythmias in Coronary Artery Disease
Other than hypertensive heart disease, coronary artery disease (CAD) is the most common cause of structural heart disease in the United States. Most patients who experience life-threatening arrhythmias have underlying structural heart disease, and the majority of patients presenting with sustained ventricular arrhythmias have underlying CAD.1 Patients with coronary disease may also have various less severe forms of arrhythmias, including bradyarrhythmias and supraventricular and ventricular arrhythmias.
Arrhythmias Associated with Acute Ischemia and Myocardial Infarction
Acute myocardial ischemia (AMI) usually results from partial or total coronary occlusion with a subsequent imbalance between myocardial oxygen supply and demand. Acute coronary syndromes (ACS) include myocardial infarction (MI) (ST-segment elevation and depression, Q wave and non–Q wave) as well as unstable angina.2,3 ACS may result in arrhythmias during acute coronary occlusion, reperfusion, myocardial infarct evolution, or the healing phase after infarction. Ischemia-induced changes in ions, metabolites, ion channels, gap junctions, and cellular and tissue architecture result in profound changes in the electrophysiological properties of the affected myocardium, which interact with modulating factors (autonomic nervous system, electrolytes, ischemic preconditioning, changes in heart rate) and the presence of concurrent structural heart disease (scar, hypertrophy, depressed ejection fraction), leading to generation of cardiac arrhythmia.
Mechanisms
Acute ischemia following coronary artery occlusion results in local tissue hypoxia and a loss of function of the adenosine triphosphate (ATP)-dependent sodium-potassium (Na+-K+) pump. Cellular membrane permeability is altered, pH falls, and a net K+ leakage from the myocyte and a rise in extracellular K+ occur. The normal cardiac resting membrane potential decreases from −80 mV to around −50 mV.4,5 The action potential (AP) amplitude falls, and maximal upstroke velocity (dV/dt max) decreases.6 Within the first 2 minutes, the fall in resting membrane potential results in an increase in conduction velocity.7 As the AP upstroke velocity falls over the next 10 minutes, conduction velocities decrease by up to 50%.8 Importantly, the effects of ischemia on the electrophysiological properties of myocardial cells are heterogeneous. AP duration and upstroke velocity are more reduced in subepicardial cells than in subendocardial cells.9 Within the central zone of ischemia, refractory periods are prolonged, and conduction velocity is decreased.10 In the surrounding nonischemic tissue, the refractory period may become shortened, and the conduction velocity may increase—possibly as a result of local catecholamines, circulating catecholamines, or both. Intermediate or mixed changes occur in the border zone between ischemic and nonischemic tissues, which results in a marked heterogeneity of electrophysiological properties. Changes in the degree of cell-to-cell coupling and tissue architecture also cause slowing and then failure of electrical propagation. The extracellular compartment shrinks, and extracellular resistance increases.11 Gap junction disruption results in cellular uncoupling. Heterogeneities in the changes in intracellular and extracellular resistance are particularly pronounced in the border zone.
Autonomic nervous system changes occur during MI. While infarcted areas show sympathetic denervation, surrounding and distant areas develop hyperinnervation.12 In addition, subepicardial sympathetic fibers traveling from base to apex may be damaged by transmural infarctions, which results in denervation of areas located more apically; these areas may show denervation hypersensitivity to catecholamines.13–18 Autonomic and neurohumoral influences can thus modify the electrophysiological properties of the substrate and can also result in arrhythmic triggers (premature ventricular contractions) via enhanced automaticity or after-depolarizations.
If the acute ischemia resolves, further myocardial injury occurs during reperfusion; this includes vascular damage, myocardial stunning, and further necrosis, mediated by intracellular calcium overload and oxygen free radicals. Calcium-dependent arrhythmias resulting from triggered activity, such as delayed after-depolarizations, may develop. Premature ventricular contractions (PVCs) and accelerated idioventricular rhythms are the most common rhythms associated with this phase, and they do not portend an adverse prognosis.19,20
Recurrent ischemia may result in alterations of the cellular metabolism and local biochemical environment and thus modulate arrhythmogenicity. Short, repetitive coronary occlusions have been shown experimentally to result in decreased incidence of ventricular fibrillation (VF) during reperfusion—a phenomenon termed ischemic preconditioning.21
Electrolyte abnormalities, particularly hypokalemia and hypomagnesemia, can alter myocardial electrophysiological properties and can also generate arrhythmia triggers. Circulating fatty acid levels have also been associated with an increased risk of sudden death as a manifestation of coronary disease.22,23
Genetic factors may play a significant role. In two retrospective studies, MI patients who experienced VF or sudden death were more likely to have a family history of sudden cardiac death (SCD).24,25 Candidate genes include genes that predispose to the development of the underlying substrate (coronary disease as well as acute plaque rupture, thrombosis, or both) and genes that directly influence the electrical properties of the myocardium and its vulnerability to ventricular fibrillation. A number of monogenic arrhythmic disorders have been well characterized (long QT syndromes, short QT syndromes, Brugada syndrome, catecholaminergic polymorphic VT syndromes).26 In addition, several genetic variants (polymorphisms) have been associated with sudden death or arrhythmia in general populations (SCN5A gene, β2-adrenergic receptor gene27,28). These genetic abnormalities and variants, and others still undiscovered, clinically apparent or subclinical, are likely to influence an individual’s susceptibility to develop arrhythmias during both acute and chronic coronary ischemia.
Stages of Ventricular Arrhythmogenesis Following Coronary Artery Occlusion
Two distinct phases of arrhythmogenesis (Table 57-1) occur during the initial 30 minutes (“acute phase”) of ischemia after experimental coronary artery ligation.8 Phase 1a arrhythmias occur 2 to 10 minutes following coronary artery occlusion (peak 5 to 6 minutes) and are caused by re-entry within the ischemic myocardium resulting from the inhomogeneity of refractory periods in normal and ischemic tissue. Mapping studies have revealed the presence of low-amplitude fractionated electrograms.7,8
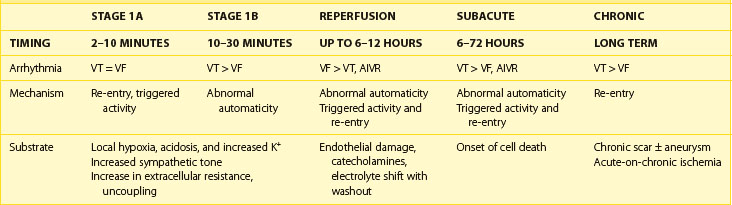
Phase 1b arrhythmias occur 10 to 30 minutes following coronary artery occlusion (peak 15 to 20 minutes). The precise mechanism of type 1b ventricular arrhythmias is unclear. By this stage, the inhomogeneities in subepicardial refractoriness and conduction have improved to near-normal values.8 Because of the important role of catecholamines in arrhythmogenesis, it has been postulated that abnormal automaticity is the underlying mechanism.29 Myocardial stretch mechanisms have also been implicated in the generation of abnormal automaticity.30 Studies in canine hearts during the first 30 minutes following coronary artery occlusion have suggested that up to 60% of ventricular tachycardias (VT) are focal in origin, arising from Purkinje fibers.31 Generally, during these two phases, spanning the first 30 minutes following occlusion, no permanent structural damage occurs. On reperfusion, ischemic cells survive and generally recover function. However, toward the end of phase 1b, changes in the internal axial resistance of cardiac tissue are first noted, indicating the onset of irreversible cellular and gap junction damage.32
The subacute or delayed phase occurs 6 to 72 hours following coronary artery occlusion (peak 12 to 24 hours).33 It coincides with the onset of cell death; reperfusion at this stage does not reduce the amount of cell damage. Although substantial myocardial cell death occurs in the infarcted region, subendocardial Purkinje fibers survive with altered electrophysiological properties predisposing to arrhythmia generation.34 A reduced resting membrane potential and spontaneous membrane depolarizations lead to abnormal automaticity. Delayed after-depolarizations resulting in triggered activity have also been demonstrated.35 In addition, heterogeneity of conduction and refractoriness at the border zone, which is the interface between the dead myocardium and the still-viable myocardium, may lead to re-entrant arrhythmias.
Reperfusion Arrhythmias
Reperfusion arrhythmias are more common after short ischemic episodes than after long ischemic periods.36 In the canine model, reperfusion arrhythmias have been shown to occur in two stages. Immediately following restoration of perfusion after coronary artery occlusion, VF may occur due to multiple wavelet re-entry. This occurs as a result of a rapid but inhomogeneous return of APs to previously unexcitable cells within the ischemic zone and a shortening of refractory periods in the border zone brought about by the washout of K+ and metabolites from the extracellular space.37 In addition, premature depolarizations may be induced by triggered activity. Although overall electrical function can return to normal at this stage, gap junction injury may persist with a corresponding inhomogeneous delay in conduction properties.
Accelerated idioventricular rhythms are commonly seen following reperfusion in the canine model. This arrhythmia may be due to the increased adrenergic stimulation of Purkinje fibers near the ischemic region causing enhanced automaticity or triggered activity.38 As accumulation of catecholamines is required, these arrhythmias typically occur after 20 to 30 minutes of occlusion. Compared with the canine model, the incidence of early reperfusion arrhythmias in the human population is significantly lower. This probably reflects the longer occlusion times and less rapid or incomplete reperfusion typically seen in patients presenting with AMI.
Clinical Characteristics of Ventricular Arrhythmias in Acute Coronary Syndromes
Ventricular arrhythmias are present in 64.1% of patients following acute ST-segment elevation myocardial infarction (STEMI).20 More than 10 PVCs per hour may be seen in 19.7% and nonsustained VT (NSVT) in 6.8% of patients. Sustained VT or VF occurs in 10.2% of admissions, with an incidence of 1.9% within the first 24 hours and 3.7% to 4.4% in the first 48 hours.39–42 Older age, systemic hypertension, previous MI, Killip class, anterior infarct, and depressed ejection fraction are associated with a higher risk of sustained VT and VF.39 Ventricular arrhythmias are more common in patients with signs of extensive left ventricular damage. However, early mortality is increased in patients who develop VT and fibrillation, even in the absence of congestive heart failure and hypotension. The incidence of VF in AMI seems to have declined over the last 20 years, whereas the incidence of VT has not changed much.43
Ventricular arrhythmias also occur in the setting of unstable angina (UA) or non–ST-elevation MI (NSTEMI), both during episodes of pain and when patients are pain free.44 In a pooled analysis of over 25,000 patients with UA or NSTEMI from four trials, the incidence of sustained VT or VF was 2.1%.45
Premature Ventricular Contractions
PVCs are seen in the majority of cases of acute MI. Early PVCs (within the first 48 hours) do not appear to affect the prognosis, but frequent or complex PVCs occurring beyond 48 hours after AMI may be associated with increased arrhythmic risk. In the human heart, R-on-T PVCs are rarely observed, accounting for only 1.8% of PVCs during the first 24 hours of admission, and most PVCs do not trigger severe ventricular tachyarrhythmias.46–48 However, in a canine model, 24% of PVCs occurring between 12 and 30 minutes (phase 1b) resulted in R-on-T and were responsible for the initiation of 34% of spontaneous episodes of VT and fibrillation.
Several studies from the prethrombolytic era have suggested that frequent PVCs (>10 PVCs per hour), complex PVCs (ventricular bigeminy, couplets, or multiform ventricular premature beats), or both are a risk factor independent of the degree of myocardial damage and left ventricular systolic dysfunction,20,49 but in another trial, PVC frequency had no independent predictive value in multivariate analysis.50 Antiarrhythmic suppressive therapy (lidocaine) has not been shown to improve outcomes, and class Ic antiarrhythmics may increase mortality. Electrophysiology study for risk stratification is currently not recommended for either early or late post-MI PVCs.
Accelerated Idioventricular Rhythm
Accelerated idioventricular rhythm (AIVR) is commonly witnessed in the first 12 hours after admission for AMI. Although more common in patients with successful reperfusion therapy, it is not a specific marker, with 63% of patients with occluded arteries still demonstrating the arrhythmia.51 The presence of AIVR does not affect the prognosis.
Nonsustained Ventricular Tachycardia
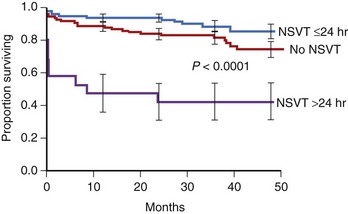
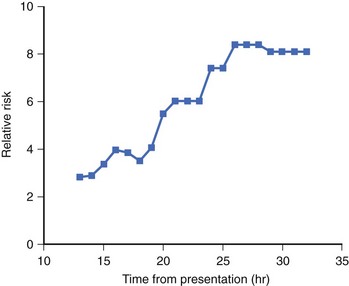
The presence of NSVT identifies patients at risk of in-hospital cardiac arrest. NSVT that occurs within the first 2 to 3 hours does not carry an adverse prognosis, whereas NSVT that occurs beyond several hours after admission does, particularly in patients with prior MI. NSVT in the setting of AMI occurs in 1% to 7% and possibly in as many as 75% of patients (Figure 57-1).52 NSVT occurring 24 hours after AMI carries a worse prognosis than NSVT occurring within the first 24 hours following AMI (Figure 57-2). This is contrary to the commonly held belief that arrhythmias occurring within the first 48 hours following MI do not carry an adverse long-term prognosis. NSVT in the setting of healing MI (7 to 10 days following MI) is also associated with a poorer prognosis. Aside from β-blockers, antiarrhythmic therapy is not currently recommended for either early or late post-MI asymptomatic NSVT. Electrophysiology testing is not currently recommended for risk stratification of NSVT in the first several weeks after AMI but is considered “reasonable” for risk stratification in patients with remote MI, NSVT, and left ventricular ejection fraction (LVEF) 40% or less.53
Ventricular Tachycardia, Polymorphic Ventricular Tachycardia, and Ventricular Fibrillation
The incidence of documented “early” sustained monomorphic VT (SMVT) within the first 48 hours of AMI is in the range of 2% to 3% in STEMI and less than 0.9% in NSTEMI.39,45,54 It may indicate extensive myocardial damage and serve as an independent predictor of mortality.54,55 As discussed above, arrhythmia mechanisms undergo dynamic changes in the early minutes and hours after onset of ischemia and may involve both re-entry (within ischemic areas of slowed conduction and increased refractoriness) or non–re-entrant mechanisms (triggered activity or increased automaticity). SMVT, however, implies stability of the ventricular depolarization pattern, which is most readily provided by a stable re-entry circuit. Thus, it is likely that SMVT, even in the early hours following MI, occurs in the presence of an already established permanent substrate (developing necrosis or pre-existing scar). Electrolyte abnormalities or ischemia that can cause the events that initiate re-entry (PVCs, NSVT) should be corrected; however, SMVT should be addressed as it would be even in the absence of these factors. From the currently available data, it is unclear that SMVT can be lumped together with other early post-MI arrhythmias in terms of its effect on the long-term prognosis—as most studies have not analyzed it separately from VF, polymorphic VT, or NSVT. In the GUSTO-I (Global Utilization of Streptokinase and t-PA for Occluded Coronary Arteries) study, patients with early (<48 hours) sustained VT had a 7.1% 1-year mortality among 30-day survivors, compared with 6.1% in patients with “late” (>48 hours) VF, and 2.6% in patients without any early or late VT or VF.39 Therefore, SMVT, occurring even “early” after MI, is generally considered by many experts to be an indicator of high risk for future arrhythmia and SCD, warranting further investigation and intervention.53,56
Polymorphic VT, which occurs in 0.3% to 2% of patients, may be a marker of ongoing ischemia; therefore, it can often be effectively managed by anti-ischemic interventions. It is more often seen in patients who also develop VF.57 In general, efforts are made to correct potential triggering factors such as hypokalemia, hypomagnesemia, abnormal serum calcium, or bradycardia (in those with bradycardia or pause-dependent onset).53 In a case series of 11 patients with polymorphic VT, none had sinus bradycardia, but 3 of 11 had a sinus pause preceding the onset.58 None had prolonged Q-T interval, hypokalemia, or abnormal serum magnesium or calcium. Nine of eleven had signs of recurrent ischemia immediately before arrhythmia onset. VF occurs in 3.7% of all acute STEMIs in the first 48 hours, and this is likely an underestimation, as prehospital events are not included.39,41 Of these, most VF episodes occur within the first 4 hours (3.1%).41 When all VF events, before and after 48 hours, were included, VF was found to occur in 6.7% of STEMI patients and in 1.3% of NSTEMI patients.39,45 In the first 4 hours of admission, VF was more likely to occur in the setting of hypokalemia, low blood pressure, larger infarct size, current smoking, and a younger age. VF was more common in inferoposterior infarcts, possibly because of greater autonomic upset. The association of initial bradycardia with early fibrillatory risk fits with the observation that vagal overactivity may precede VF. VF at all stages of infarct evolution is more common in patients with larger infarcts as determined by serial cardiac enzyme measurements.59
Traditionally, primary VF has referred to VF that occurs during the first 48 hours of an uncomplicated MI (without recurrent ischemia or heart failure), and in the GISSI (Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico)-2 trial, it was associated with increased in-hospital mortality; however, no statistically significant association with post-discharge mortality (1-year mortality of those who survived to hospital discharge) was observed. These effects of early ventricular arrhythmias on early mortality were confirmed in the percutaneous coronary intervention era.60,61 In contrast, nonprimary VF (VF occurring in the setting of recurrent ischemia or heart failure or beyond the first 48 hours following MI) was associated with marked increases in both 30-day mortality and 6-month mortality.41 In the GUSTO trial, all “late” (>48 hours following MI) sustained VT, VF episodes, or both were associated with markedly increased long-term mortality at 1 year among 30-day survivors.39 More recently published data from the GUSTO-V trial found that “early” VT or VF (<48 hours) was associated with increased in-hospital mortality but not with 1-year mortality among 30-day survivors42; however, all arrhythmias (VF, all VT) were pooled in this analysis. The temporal cutoff between “early” and “late” arrhythmias at 48 hours following MI is arbitrary to some extent; data to suggest that this should be at 24 hours or even earlier exist52; clearly, decisions should be individualized and based on expert evaluation and judgment. Moreover, additional tools for risk stratification are needed, and this is an area of active investigation; in the future, these may include a combination of electrophysiological testing, genetic evaluation, scar imaging, autonomic evaluation, and so on.
Preliminary data from a post hoc analysis suggest that ranolazine, an antianginal that inhibits the late inward Na+ current, may decrease the incidence of VT or VF (as well as SVT or AF), but this requires further study.62 A large pooled analysis has suggested that early administration of intravenous β-blockers in acute myocardial infarction may decrease the mortality and incidence rates of VF, but the Clopidogrel and Metoprolol in Myocardial Infarction Trial (COMMIT) performed on 46,000 patients with AMI did not show the mortality benefit of this intervention.63,64
In summary, the available data on the outcomes of ventricular arrhythmias in AMI have limitations. These data come mostly from thrombolytic trials, retrospective analyses, limited numbers of events, and analyses of multiple types of arrhythmia, rather than specific arrhythmias, pooled together. Mainly on the basis of large thrombolytic trials, “early” sustained VF or polymorphic VT occurring within the first 24 to 48 hours of an uncomplicated AMI is associated with increased in-hospital and 30-day mortality but appears to have little effect on long-term mortality in patients surviving hospital discharge; however, this may be because high-risk patients die during their initial hospital stay. Conversely, all sustained ventricular arrhythmias that are “late” (>24 to 48 hours following MI) or in the context of complicated MI and any sustained monomorphic VT are considered indicators of high risk of arrhythmia and SCD, and patients are considered survivors of cardiac arrest. The temporal cutoff between “early” and “late” arrhythmias is unclear and may be close to 24 hours or even earlier. Occasionally, complete revascularization may be achievable with sufficient treatment—in the absence of prior MI, residual scar, SMVT, or systolic dysfunction—but most of these patients should be considered for defibrillator implantation, and expert, individualized decisions should be made. Detailed acute and chronic management guidelines for ventricular arrhythmias associated with AMI have been published.2,3,53,56
Supraventricular Arrhythmias in the Setting of Acute Ischemia
Incidence
Holter monitoring has revealed that 15% of patients recovering from MI have supraventricular tachycardia, ranging from single atrial premature beats (APBs) to sustained AF during their hospital stay.65 The prevalence of SVT increases during the first month after MI. Overall, the incidence of AF in AMI in the modern era is 7.8% to 28%.66–71
Mechanism of Atrial Fibrillation During Acute Myocardial Ischemia
The pathophysiology of AF that occurs in the course of AMI has many components. Inflammation (pericarditis), changes in hemodynamics (atrial stretch and dilation), and atrial ischemia may all play a role.72–75 Following significant ventricular damage, end-diastolic volume and pressure rise, causing an increase in atrial pressure and wall tension. This predisposes to AF and also explains the close relationship between heart failure and AF in the setting of MI. In an angiographic study, AF that occurred during inferior MI was shown to more likely occur in the setting of an occluded proximal left circumflex artery, with or without right CAD, if it was combined with impaired perfusion of the AV nodal artery.76 AF did not occur in patients with right coronary artery occlusions if the circumflex artery was unobstructed. In a series of 266 patients, all 12 who developed atrial arrhythmias had inferior infarction. In the vast majority of these patients, the sinus node artery was distal to the site of right coronary occlusion, which suggests that sinus node ischemia may also play a role.77 Evidence of atrial infarction in the 12-lead electrocardiogram (ECG; manifesting as PR-segment displacement) may also predict the onset of AF during AMI.78 Other risk factors include advanced age, the presence of congestive heart failure, three-vessel coronary disease, right coronary artery (RCA) occlusion, female gender, anterior Q-wave MI, previous MI, and previous coronary artery bypass graft (CABG).66,79
Consequences of Atrial Fibrillation During Acute Myocardial Infarction
The development of AF results in the loss of atrial contraction and rapid, irregular heart rates, which, in turn, will cause impaired diastolic filling and increased myocardial oxygen demand. Atrial contraction is an important component of ventricular filling, particularly in failing hearts. In the ischemic canine heart, induced AF was shown to cause a reduction in cardiac output, a fall in mean aortic pressure, and a fall in mean myocardial blood flow.80 This may precipitate a vicious downward spiral, with AF exacerbating heart failure, which, in turn, promotes AF. Both will increase the ischemic burden and the likelihood of ventricular arrhythmias.
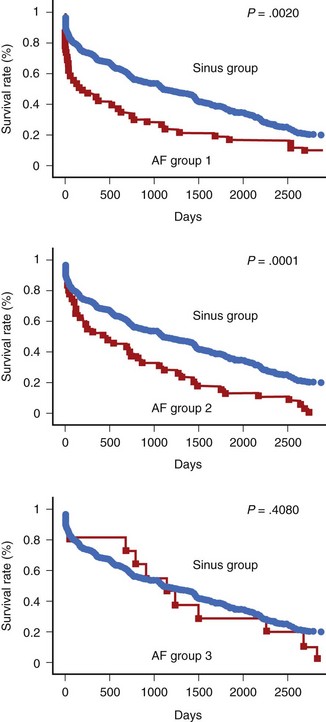
In patients who sustain an AMI, hospital mortality is significantly higher in those with AF than in those without it (Figure 57-3).66,68,69,71,72 It has been suggested that AF may be a risk factor for VF.81 AF occurs in patients with signs of heart failure and larger infarctions. In large-scale trials, the negative impact of AF has been shown to be independent of other variables.70,79 However, it is possible that the increase in in-hospital mortality is restricted to those patients with new-onset AF (after admission) rather than pre-existing AF (see Figure 57-3).70,82
Bradyarrhythmias in the Setting of Acute Ischemia
High-degree atrioventricular (AV) block is seen in a significant proportion of patients presenting with acute inferior MI. The incidence of advanced (second-degree and third-degree) AV block in the thrombolytic era ranges from 5.6% to 3.7% of all AMI patients.83 In inferior wall MI, the reported incidence ranges from 7.3% to 9.8% of patients developing advanced AV block to 13% of patients having complete heart block, compared with 3.2% advanced AV block in patients with acute anterior wall infarction.84–86
All studies examining patients with heart block after infarction have found an association with a greater degree of myocardial damage, whether measured by cardiac enzymes, echocardiography, or nuclear scintigraphy.85,87–90 As it has long been recognized that heart block is most prevalent in patients with inferior wall MI (two- to threefold increase compared with anterior AMI patients), the majority of studies were done in this population of patients.84,86 Within this group, right ventricular involvement also appears to be associated with the development of advanced AV block.88
Patients with inferior MI and coexisting left anterior descending coronary artery obstruction have a sixfold greater chance of developing heart block in the acute phase of infarction than do patients with inferior infarction without such obstruction.91 The site of left anterior descending artery occlusion is usually proximal to the origin of the first septal perforator. These findings suggest that the proximal AV conduction system has a dual arterial blood supply from both the right and left anterior descending coronary arteries and may explain the transient behavior of heart block and lack of necrosis of the AV node seen in many patients with inferior MI. A histopathologic study of hearts with posteroinferior MI has shown a strong correlation with atrial infarction in the region of the inputs to the AV node but a lack of correlation with infarction of the specialized conducting system.92
In the setting of inferior infarction, patients with CHB have higher mortality; more episodes of VF or tachycardia; and sustained hypotension, pulmonary edema, pericarditis, and atrial fibrillation than do patients without heart block.71,84,85,89,90 In contrast, in those with inferior MI who survive to hospital discharge, the presence of heart block has no effect on long-term mortality.84,85,89
Differences between patients who develop heart block early and those who develop it late in the course of their AMI do exist. Different studies, however, reveal conflicting data. Sclarovsky et al reported that patients who develop early advanced block—defined as that occurring with continuing hyperacute changes of AMI on the ECG—had CHB that was of short duration, was unresponsive to atropine, and often required pacemaker therapy.93 Symptoms of syncope, heart failure, and cardiogenic shock were frequently present. Patients with late block typically had second-degree heart block of longer duration, had a positive response to atropine, and rarely required pacemaker therapy. The mortality rate was high in the early group (23%) compared with that of the late group (7%). In another study, using a 6-hour cutoff time limit from admission, patients with inferior MI were divided into those who developed second- or third-degree block early and late.94 In the early group, all patients had transient AV block that appeared suddenly, disappeared by 24 hours, and displayed a positive response to atropine. In the late group, heart block was often preceded by first-degree block, lasted longer, had a relatively fast ventricular escape rhythm, and had little response to atropine. A third study, dividing patients on the basis of AV block appearing before or after 24 hours from admission, found no significant difference in hospital mortality.95
The mechanisms responsible for AV block during acute inferior MI would, therefore, appear to be multiple and related to the time course. Along with acute necrosis of the perinodal atrial myocardium or specialized conduction tissue, increased parasympathetic tone is a factor that is usually postulated; however, persistence of AV block after atropine administration is frequently observed. It has been demonstrated that endogenously released adenosine in the oxygen-deprived myocardium can cause AV block.96 Thus, not surprisingly, it has been reported that aminophylline may be successful in restoring sinus rhythm in atropine-resistant patients with inferior infarction.97–99
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


