Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a disorder characterized clinically by ventricular arrhythmia, heart failure and sudden death, and histologically by cardiomyocyte loss and replacement with fibrous or fibro-fatty tissue. The estimated prevalence of ARVC is 1 in 5000 of the population. It is an important cause of sudden cardiac deaths in athletes and in people under 35 years. In many individuals, the disease is caused by mutations in genes that encode different components of the intercalated disc of cardiomyocytes. Clinically, ARVC is difficult to diagnose, usually requiring integration of data from family members, electrocardiography and a range of imaging techniques. Management of ARVC focuses on treatment of symptomatic arrhythmia, prevention of sudden cardiac death and, in the later stages of the disease, heart failure management.
In 1952 Dr Henry Uhl described a case of “almost total absence of the myocardium of the right ventricle” in an 8-month-old girl.1 The case was characterized by complete heart block and right ventricular failure. At post mortem the myocardium of the left ventricle was normal and there was a sharp demarcation line between this and the very abnormal right ventricle in which there was virtually complete loss of the myocardium. There was no evidence of ischemic change or inflammation and the cause was assumed to be a maldevelopment or dysplastic process.
The first description of an ARVC was in 1978, when detailed electrocardiography readings of four French patients with right ventricular dysplasia associated with arrhythmia were reported.2 In the same journal there was a report of two French patients in their sixth decade of life presenting with recurrent ventricular tachycardia of left bundle branch bock (LBBB) morphology resistant to medical therapy. Clinical examination, angiography and ultimately post-mortem examination identified what was described as “the parchment right ventricle syndrome of the adult.”3
A case series of 24 patients published 4 years later identi-fied the characteristics of the typical patient with what is now called arrhythmogenic right ventricular cardiomyopa-thy. The mean age at presentation (in those who had symptoms) was 39 years, and the male/female ratio was 2.7:1. Almost 90% of the patients had T-wave inversion in the right precordial leads. Ventricular postexcitation waves (epsilon waves) were present in one-third of patients. All but one of the patients had spontaneously occurring ventricular tachycardia with a LBBB configuration. In those who underwent echocardiography, the ratio of right to left ventricular size was increased, and wall motion abnormality of the right ventricle was seen in the majority. One patient had a wall motion abnormality of the posterior wall of the left ventricle. Morphologic findings were confirmed in 13 patients during surgical intervention. The right ventricle was invariably enlarged and there were discrete areas of segmental dilation associated with dyskinetic wall motion and aneurysms. The authors reported the most common location of these right ventricular aneurysms to be the anterior surface of the right ventricular outflow tract, apex and inferior wall, and coined the term “the triangle of dysplasia.” There was frequently an increase in subepi-cardial fat over these areas and depletion in the numbers of muscle fibers. The amount of fibrosis was variable. On microscopy some hypertrophy of the remaining myofibers was noted, and there was a varying degree of lymphocytic infiltration.4 One of the patients came from a family reported 4 years earlier in a description of seven cases of familial ventricular tachycardia.5
The first gene mutation to be associated with ARVC was identified in patients with a recessive cardiocutaneous syndrome characterized by wooly hair, palmoplantar kerato-derma and ARVC common on the Greek island of Naxos.6 The locus for Naxos disease was mapped to chromosome 17 (17q21), and subsequently a deletion in the gene encoding plakoglobin was identified as the disease-causing mutation.7,8 Plakoglobin is a constituent protein in both adherens and desmosomal cell–cell junctions. Subsequent evaluation of other related desmosomal genes in ARVC patients with more typical autosomal dominant disease has identified disease-causing mutations in a further four genes encoding other desmosomal proteins.
The structure of the right ventricle renders it inherently more difficult to image than the left ventricle, but improvements in imaging techniques over the past decade have broadened the clinical spectrum of the disease by identifying patients at an earlier, often asymptomatic stage. Cardiac magnetic resonance imaging in particular has demonstrated a high prevalence of left ventricular involvement in many patients, illustrating the heterogeneity in disease expression.9–13
In most populations the prevalence of ARVC is unknown. An apparent cluster of disease is reported in the Veneto region of Italy, with a frequency of 1 per 5000, but it is unclear whether this is a true clustering or a reflection of local interest and expertise in the disease. Nevertheless, the limited information available suggests that ARVC is an important cause of premature sudden cardiac death. A retrospective review of a French post-mortem series identified ARVC as a likely cause of death in 10% of the population aged 1–65 years.14 Prospective studies of an Italian cohort aged 12–35 years found evidence of ARVC in 12% of those who suffered a sudden cardiac death.15 A population-based study from the USA identified pathologic changes of ARVC in 17% of sudden death victims under 40 years of age.16 Other authors report that up to 25% of sudden deaths in athletes are due to ARVC.17
In the early stages, abnormal pathologic findings are localized to the apical, inflow and infundibular areas of the right ventricle.4 With disease progression, the left ventricle, particularly the posterolateral wall, is often involved, with relative sparing of the interventricular septum. In some cases, the pathologic abnormalities may be confined to the left ventricle.18,19
Macroscopic examination of the heart shows diffuse or focal thinning of the right ventricular wall, affecting the subepicardium more than the subendocardium, with aneu-rysm formation in up to 50% of cases.18,20 Histologically, there is myocardial atrophy and fibro-fatty replacement. Islands or strands of surviving myocytes exhibit a combination of degenerative change with myocyte vacuolation, often associated with focal mononuclear inflammatory cell infiltrates.21–24 In some cases, typical histologic findings may be present in the absence of any macroscopic features.25 A second histologic pattern, characterized by trans-mural infiltration of adipose tissue without fibrous replacement or wall thinning, has been considered to be an earlier stage of the condition22,26 but this can be a normal finding, particularly in older women.20
A number of studies have reported evidence of apoptosis in ARVC which is not found in normal controls, patients undergoing post-transplant surveillance or in patients with structurally normal hearts and VT arising from the RV outflow tract.27–30 Apoptosis is more common in patients with a short (less than 6 months) clinical history and acute symptoms and signs.27 Apoptosis is present to an equivalent degree in children and adults with ARVC.28
Endomyocardial biopsies from some ARVC patients have been found to contain enteroviral RNA with homol-ogy to Coxsackie virus type B. In one study similar proportions of patients with myocarditis or dilated cardiomyopathy were positive for viral RNA but none of the control patients with non-inflammatory cardiac disorders.31 Other studies have failed to confirm these findings, which may reflect patient selection and differences between inherited and sporadic, non-familial forms of ARVC.32–37
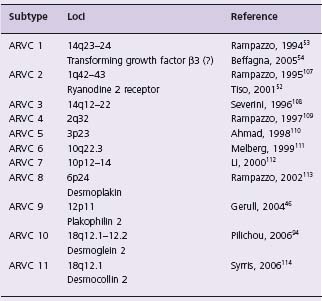
Systematic family studies have shown that ARVC is inherited in up to 50% of cases.38 Numerous genetic loci have been identified (Table 45.1). The mode of transmission is usually autosomal dominant with variable penetrance, but recessive forms are well recognized and provided the first insights into the genetic basis of ARVC. In 1986, an autosomal recessive syndrome characterized by cardiomyopathy, wooly hair and palmoplantar kerato-derma was described in families from the Greek island of Naxos.6 The cardiac phenotype of this syndrome replicates the clinical and histopathologic characteristics of typical ARVC.39 Subsequent molecular genetic investigations in families with Naxos disease revealed a homozygous mutation in the gene encoding plakoglobin, a member of the armadillo protein family found in adherens and desmo-somal junctions between cardiomyocytes. Families with a similar cardiocutaneous phenotype are reported from other Mediterranean areas,40–43 India44 and South America.45 The cause in many of these families is an autosomal recessively inherited mutation in the gene encoding desmoplakin.
The intercalated discs are end-to-end connections between cardiomyocytes that maintain mechanical and electrical integrity between cells. They consist of three main components: the adherens junction, the desmosome, and gap junctions. Desmosomes are the secondary mechanical intercellular junctions, present in abundance in epithelial tissues and the myocardium. Plakoglobin links the intermediate filament network to the cytoplasmic tails of the cadherin proteins which extend into the intercellular space and have adhesive functions, although the precise mechanism by which they achieve this function is unknown. Des-moplakin is a protein of the plakin family of cytolinkers and is located deeper in the cytoplasmic plaque of the desmosome. It appears to be an obligate component of the desmosomal junction.37
The involvement of plakoglobin and desmoplakin genes in these recessive forms of ARVC led to the examination of these and other desmosomal protein genes in the more common dominant ARVC phenotype. Plakoglobin has only been associated with autosomal recessive ARVC to date. Desmoplakin, on the other hand, accounts for 16% of dominant ARVC cases. Genes implicated only in autosomal dominant disease include plakophilin 2 (PKP2) accounting for 25–43% of cases,46–48 desmoglein (DSG2) (10% of cases), and desmocollin (DSC2).
The discovery that many cases of ARVC are “desmo-somal” diseases has led to a simplistic theory of pathogen-esis in which disruption of desmosomal integrity causes separation of myocardial cells under conditions of mechanical stress. Detached myocytes die and are replaced by fibro-fatty tissue. The predilection for the right ventricle can be explained by the relative thinness of the RV walls in comparison with the left ventricle; the tendency for lesions to develop in the “triangle of dysplasia” is explained by the fact that the RV inflow, apex and outflow are the regions of maximal mechanical stress.
While this theory may explain the ultrastructural changes associated with ARVC and, indeed, the apparently aggressive form of the disease in those who partake in endurance training,49 it does not account for the disproportionate tendency to arrhythmia. Evaluation of a small number of patients with Naxos disease has shown that expression of connexin 43, a major gap junction protein, is markedly reduced in both the left and right ventricles, even at an early age of disease development. A reduction in the number and size of the gap junctions may result in an electrical coupling defect which increases the propensity to arrhythmia and may explain the occurrence of sudden death in patients without marked morphologic changes.50
Two non-desmosomal genes have been associated with ARVC. Mutations in the ryanodine receptor gene (RYR2) were reported in association with a localized form of ARVC, but mutations in this gene more typically cause catechol-aminergic polymorphic VT (CPVT) and are no longer clas-sified as a subtype of ARVC.51,52 A mutation in the transforming growth factor beta 3 (TGFβ3) gene has also been described, but the mutation occurs in an intronic region of the gene and only one of the three originally described ARVC1 families has a TGF β 3 mutation. Functional studies have shown an alteration in the expression of the TGFβ3 gene resulting in increased fibrosis. Whether this represents true ARVC or not remains controversial.53,54
The natural history of ARVC can be divided into four phases.38 In phase one (the “concealed” phase) patients are asymptomatic with few if any morphologic abnormalities. In the absence of systematic post-mortem evaluation of the right ventricle, the diagnosis may be missed at this stage and so the incidence of sudden death in this phase may be underestimated. In the second (electrical) phase, symptoms of arrhythmia occur and morphologic structural and functional abnormalities are easier to identify. The third phase is marked by the classic right ventricular functional abnormalities that can progress to right ventricular failure with relatively preserved left ventricular function. The final or end-stage is associated with the development of biven-tricular systolic impairment. Although a useful disease paradigm, it is not inevitable that affected persons will progress through all phases as described. Moreover, multiple (mostly unknown) factors influence the clinical manifestations and natural history of the condition. It is generally thought that physical training and exercise both increase the risk of arrhythmic events in affected individuals and result in more aggressive disease progression.17,49,55–57 Some patients demonstrate left ventricular involvement early in the disease, and may have predominant left ventricular disease.58–60
The first presentation of the disease is often sudden cardiac death in previously asymptomatic individuals, including young children and teenagers.14,55,61,62 Occasionally, patients will have experienced syncope in the months preceding their death.14,56,63 An imbalance of adrenergic activity has been suggested as a possible factor in the genesis of lethal ventricular arrhythmias; thus exposure to catecholamines (particularly during exercise) may increase the risk of sudden death.64
Patients usually present with symptoms between the second and fifth decades of life. Syncope, palpitations and chest pain are common, but family studies have shown that most affected individuals are asymptomatic, particularly in the early stages.65,66 With disease progression, features of right and later biventricular failure may be present.
Accurate diagnosis of ARVC is notoriously difficult at post mortem and in living patients. The gold standard for making the diagnosis is the demonstration of transmural fibro-fatty replacement of right ventricular myocardium. However, endomyocardial biopsy has a substantial false-negative rate caused by the patchy distribution of the disease and the fact that the interventricular septum, which is the site of choice for endomyocardial biopsy, is often relatively spared. In an attempt to address this difficulty, a task force of international experts was convened, under the auspices of the Scientific Council of the International Society and Federation of Cardiology (ISFC) and the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. The Task Force reviewed the evidence available at the time in order to agree a set of clinical diagnostic criteria. They grouped clinical criteria into six categories, and assigned either major or minor status to each criterion depending on the degree of specificity for the diagnosis. A cumulative scoring system was devised with agreed thresholds for making a clinical diagnosis. The presence of two major criteria from different categories, or one major and two minor, or four minor criteria, all from different categories, is considered diagnostic of ARVC67 (Box 45.1).
BOX 45.1 Criteria for diagnosis of ARVC (adapted from McKenna et al67)
1. Family history
Major
- Familial disease confirmed at necropsy or surgery
Minor
- Family history of premature sudden death (<35 years) due to suspected right ventricular cardiomyopathy
- Family history of clinical diagnosis based on the present criteria
2. Tissue characterization of walls
Major
- Fibro-fatty replacement of myocardium demonstrated on endomyocardial biopsy
3. Global and/or regional dysfunction and structural alterations
Major
- Severe dilation and reduction in systolic function of RV with no (or only mild) impairment of LV
- Localized RV aneurysms (akinetic or dyskinetic areas with diastolic bulging)
- Severe segmental dilation of the RV
Minor
- Mild global RV dilation and/or reduction in ejection fraction with normal LV
- Mild segmental dilation of RV
- Regional RV hypokinesia
4. Depolarization/conduction abnormalities
Major
- Epsilon waves or localized prolongation of the QRS complex in V1–V3 (>110ms)
Minor
- Late potentials demonstrated on signal-averaged ECG
5. Repolarization abnormalities
Minor
- Inverted T-waves in right precordial leads (V2 and V3)
- (individuals > 12 years of age; in the absence of right bundle branch block)
6. Arrhythmias
Minor
- Sustained and non-sustained ventricular tachycardia with LBBB morphology (documented on ECG, Holter or exercise testing)
- Frequent ventricular extrasystoles (>1000 over 24-hr Holter monitoring)
Imaging
Morphologic abnormalities of the right ventricle are challenging to identify in vivo, particularly when they are relatively subtle. At the time of publication of the Task Force criteria the most commonly used imaging modality was cine-angiography of the right ventricle. This has been largely replaced by transthoracic echo which, in addition to the detection of ARVC, has a major role in the exclusion of congenital heart disease such as partial anomalous venous drainage, Ebstein’ s anomaly and other causes of isolated right ventricular abnormalities.68,69 The major challenge when using echo to assess the right ventricle is the chamber’s complex three-dimensional structure. Together with the segmental nature of the disease, this means that structure and function should be assessed using multiple views.70 The echocardiographic features of ARVC include: right ventricular dilation and hypokinesia; aneurysms; regional wall motion abnormalities, including dyskinesia of the inferobasal right ventricular segment; increased echogenicity of the moderator band; and right ventricular apical hypertrabeculation.63,68,71–73 Tricuspid annular early diastolic velocities are reduced70,74 but strain rate imaging has not yet been found to be a reliable diagnostic tool in ARVC. Three-dimensional echo may be a more accurate method of assessing right ventricular systolic function75 but larger studies are needed to validate the technique.
Cardiac magnetic resonance imaging is generally considered to be the imaging modality of choice in ARVC, but unfamiliarity with normal right ventricular morphology and function has led to high interobserver error and poor reproducibility in some series.76,77 The presence of intra-myocardial fat deposition in the right ventricle must also be interpreted with caution, as fat in the right ventricle is present in a large proportion of “normal” individuals (up to 85%), and the frequency and severity increase with age, body weight and female gender.20,23,78
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


