Aortic Valve Disease: Introduction
Aortic valve disease has traditionally been classified on the basis of valve dysfunction: stenosis versus regurgitation. Now, classification is more appropriately based on the underlying pathology because progression and management are (at least partly) determined by disease etiology, not just valve hemodynamics. Thus this chapter first describes the causes of aortic valve disease and then discusses the hemodynamics of valve dysfunction, followed by a summary of clinical outcomes, medical therapy, and timing of surgical intervention.
The prevalence of significant aortic valvular heart disease (moderate severity or worse) increases with age, present in only 0.7% of those age 18 to 44 years but increasing to 13.3% of adults 75 years and older.1 Compared with other types of clinically significant valve disease seen on echocardiography, native aortic valve stenosis was the most common (34%) in the Euro Heart Survey, followed by previous valve surgery (28%), mitral regurgitation (25%), and multivalve disease (20%).2
Normal Valve Anatomy
The normal aortic valve consists of three leaflets suspended in the aortic sinuses of Valsalva. Anatomically, the normal aortic valve has a complex three-dimensional shape, with each cusp consisting of a curved aortic sinus with the semi-cylindrical leaflet attached superiorly at the commissures and inferiorly at the junction with the left ventricular (LV) outflow tract (Fig. 76–1).3,4 The coronary ostia in each sinus give their names to the aortic leaflets: the right coronary cusp, the left coronary cusp, and the noncoronary cusp. In diastole, the leaflets overlap slightly with each other, and there is an area of central thickening at the tip of each leaflet (the nodule of Arantius), which helps to ensure complete valve closure. In systole, the leaflets open passively into a near circular orifice as ventricular contraction results in ejection of blood across the valve into the aorta. Histologically, the layers of the aortic valve leaflet are as follows (Fig. 76–2)5:
- Fibrosa—a dense collagenous layer that provides tensile strength
- Spongiosa—loose connective tissue at the base of the leaflets
- Ventricularis—elastin-rich layer on the ventricular side of the valve
- Endothelium—outer layer on both the aortic and ventricular sides of the leaflet
Figure 76–1.
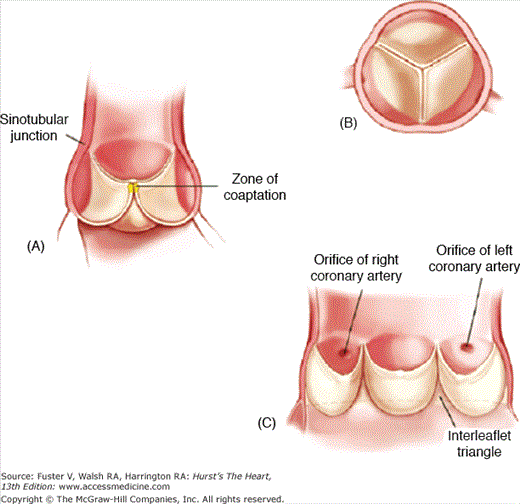
Normal anatomy of the aortic valve and aortic root. A. Long-axis cut through the aortic root. The aortic valve leaflets are suspended in the aortic sinuses of Valsalva. Each cusp consists of a leaflet attached superiorly at the commissures and inferiorly at the junction with the left ventricular outflow tract. The zone of leaflet coaptation is 1 to 2 mm with an area of central thickening at the tip of each leaflet (nodule of Arantius) to help ensure complete valve closure. B. The anatomic equivalent of a short-axis view of the aortic valve with the noncoronary cusp on top, the right coronary cusp to the bottom (left), and the left coronary cusp to the bottom (right). C. The coronary ostia in each sinus give their names to the aortic leaflets. Here, the aorta is shown opened between the right and left coronary cusps to show the relationship to the coronary ostia. Reproduced with permission from Otto and Bonow.4 Copyright © 2009, Elsevier.
Figure 76–2.
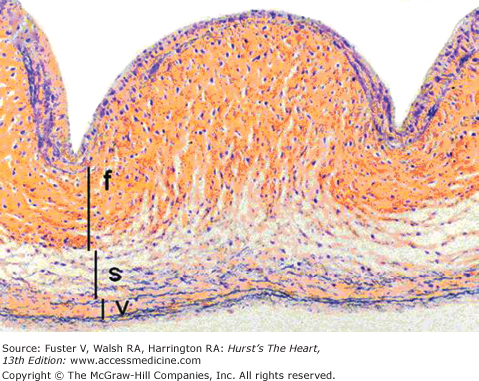
Low-magnification (×100) cross-sectional photomicrograph of an aortic valve cusp, demonstrating three layers: ventricularis (v), spongiosa (s), and fibrosa (f). Movat pentachrome stain (collagen yellow; elastin black). Reproduced with permission from Schoen and Edwards.5 Copyright © 2001, Elsevier.
In most individuals, macroscopic changes of the valve with aging include a diffuse increase in leaflet thickness and increased thickness of the central nodules. Functional systolic opening of the aortic valve is not usually affected by aging. Microscopically, there is evidence of disorganization in the fibrosa layer and increased adipose cells in the spongiosa layer. In older adults, a small amount of aortic regurgitation and mild dilation of the aorta are common, often in association with chronic hypertension.
Causes of Aortic Valve Disease
Aortic stenosis typically is due to disease of the valve leaflets. Aortic regurgitation may be due to leaflet or aortic root disease (Tables 76–1 and 76–2). The primary causes of aortic leaflet disease are a congenitally abnormal (often bicuspid) valve, calcific valve disease, or rheumatic valve disease. Abnormalities of the aorta that result in valve dysfunction include genetic connective tissue disorders, such as Marfan syndrome, or other causes of aortic root dilation, such as systemic inflammatory disorders, including systemic lupus erythematosus or rheumatoid arthritis. Metabolic disorders with associated infiltration of normal tissue (ie, Fabry disease) rarely cause aortic stenosis. Rarely, the use of some pharmacologic agents, such as serotonin inhibitors, results in leaflet thickening and valve regurgitation.
| Common |
| Bicuspid aortic valve |
| Calcific aortic valve disease |
| Rheumatic valve disease |
| Uncommon |
| Congenital aortic stenosis |
| Homozygous type II hyperlipoproteinemia |
| Metabolic infiltrative disorders (eg, Fabry disease) |
| Systemic lupus erythematosus |
| Ochronosis with alkaptonuria |
| Nonvalvular left ventricular outflow obstruction |
| Subaortic membrane |
| Hypertrophic cardiomyopathy |
| Supravalvular aortic stenosis (eg, Williams syndrome) |
| Leaflet abnormalites |
| Chronic regurgitation |
| Bicuspid aortic valve |
| Calcific valve disease |
| Rheumatic valve disease |
| Myxomatous valve disease |
| Rheumatoid arthritis |
| Nonbacterial thrombotic endocarditis |
| Systemic lupus erythematosus |
| Pharmacologic agents |
| Acute regurgitation |
| Endocarditis |
| Iatrogenic leaflet damage |
| Ruptured leaflet fenestration |
| Blunt chest trauma |
| Abnormalities of the aorta |
| Chronic regurgitation |
| Marfan syndrome |
| Bicuspid aortic valve disease |
| Hypertensive aortic dilation |
| Familial aortic aneurysm |
| Cardiovascular syphilis |
| Ankylosing spondylitis |
| Other systemic inflammatory disorders |
| Acute regurgitation |
| Aortic dissection |
Calcific aortic valve disease (CAVD) is abnormal calcification of the valve leaflets, characterized by involvement of the leaflet bases, relative sparing of the leaflet edges, and absence of commissural fusion. CAVD spans the range from mild focal areas of valve thickening without impairment of leaflet motion, called aortic sclerosis, to significant obstruction of LV outflow, called calcific aortic stenosis (Fig. 76–3). CAVD may be present on an anatomically normal trileaflet aortic valve or on a congenitally bicuspid valve. Once severe calcification is present, it may be difficult to identify the number of leaflets with certainty on clinical imaging.
Figure 76–3.

(A) In the early lesion of calcific aortic valve disease there is subendothelial accumulation of oxidized low-density lipoprotein (LDL), production of angiotensin II, and inflammation with T lymphocytes and macrophages. With disease progression, there is production of proteins that mediate tissue calcification, such as osteopontin, osteocalcin, and bone morphogenic protein 2, and leaflet fibroblasts undergo phenotypic transformation into osteoblasts. Inflammatory signaling pathways, including tumor necrosis factor (TNF) α, tumor growth factor (TGF) β, the complement system, C-reactive protein, and interleukin-1β are activated. Microscopic extracellular calcification, seen in the early lesion, progresses to areas of frank bone formation in end-stage disease. Macroscopically, there is progressive leaflet thickening and calcification (B) which leads to outflow obstruction and a corresponding increase in the Doppler aortic-jet velocity (C). Reproduced with permission from Otto.11 Copyright © 2008 Massachusetts Medical Society. All rights reserved.
Aortic valve sclerosis was present in 26% of adults older than 65 years and 48% of those older than 85 years of age in the US population-based Cardiovascular Health Study (CHS), which included 5621 adults.6 Similarly, in the Finnish Helsinki Aging Study, 21% of adults aged 55 to 71 years had aortic sclerosis on echocardiography.7 Aortic sclerosis may progress over time to stenosis. Severe calcific aortic stenosis occurs in approximately 2% to 4% of adults older than 65 years, with increasing prevalence with age. Of the approximately 50,000 aortic valve replacements performed annually in the United States, the vast majority are for aortic stenosis. On the basis of pathologic examination of surgically removed valves, approximately 50% of calcified valves removed at surgery are congenitally bicuspid and approximately 50% are due to calcification of a trileaflet valve.8
At the tissue level, the early lesion of CAVD is characterized by focal subendothelial lesions on the aortic side of the leaflet, with extension of the disease process into the adjacent fibrosa (Fig. 76–4). These early lesions contain extracellular lipid, including oxidized low-density lipoprotein (LDL), an inflammatory cell infiltrate with macrophages and T lymphocytes, and microscopic tissue calcification.9-13 There is evidence of angiotensin- converting enzyme production by a subset of macrophages, along with proteins associated with tissue calcification. Studies of surgically excised valves and animal models of CAVD suggest that disease progression involves phenotypic transformation of valvular interstitial cells, with subsequent osteogenesis resulting in cartilage and bone formation in end-stage disease (see Fig. 76–3). Ongoing research is directed toward understanding the specific molecular and cellular events involved in initiation and progression of disease.
Figure 76–4.
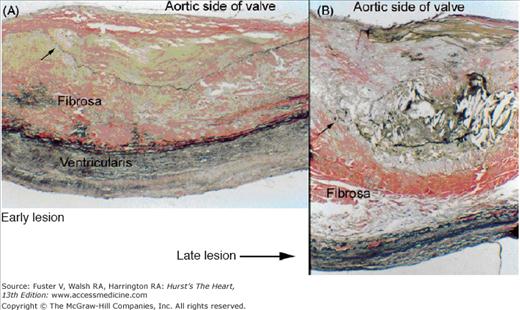
The early lesion of calcific aortic valve disease (A) demonstrates accumulation of cells and extracellular lipid and matrix in a subendothelial location on the aortic side of the leaflet, with displacement of normal subendothelial elastic lamina (arrow). In the late lesion of calcific aortic valve disease (B), there is more prominent accumulation of lipid, cells, and extracellular matrix. The elastic lamina is displaced and fragmented (arrow) (Verhoff-van Gieson stain, original magnification ×100). Reproduced with permission from Freeman and Otto.9
Development of CAVD is associated with several anatomic, genetic, and clinical factors. The strongest predictor of CAVD development is a congenitally bicuspid valve. However, genetic factors may be important, even in those with an anatomically trileaflet valve. Evaluation of genetic factors associated with CAVD is challenging because of the relatively late onset in life of clinically manifest valve dysfunction. Nevertheless, a study from France with a stable population base showed geographic clustering of aortic stenosis cases where families with multiple affected members over several generations were identified.14 Additionally, case control studies looking at specific candidate genes have shown genetic polymorphisms associated with CAVD, including variants of the vitamin D receptor, estrogen receptor, apolipoprotein E4, and interleukin 10 alleles.12,15-17
Clinical factors associated with CAVD have been examined in several large studies, including the prospective CHS study, based on echocardiographic detection of CAVD, and the Multi-Ethnic Study of Atherosclerosis (MESA), based on CT detection of leaflet calcification. In CHS, clinical factors associated with CAVD identified by multivariate analysis included older age, male sex, elevated serum lipoprotein (a) and LDL levels, hypertension, smoking status, and shorter stature.6 The MONICA-KORA study corroborated these findings, demonstrating associations with age, current smoking, and total serum cholesterol levels.18 The MESA data demonstrated additional associations with diabetes and metabolic syndrome.19 CAVD has also been associated with renal dysfunction and abnormalities in calcium and phosphate metabolism. The strength of these associations with CAVD is comparable to that seen with atherosclerotic disease (Table 76–3).6,18-20 Other supporting data for an association of CAVD with atherosclerotic risk factors is the presence of accelerated CAVD in the aortic sinuses and root seen early in life in patients with homozygous type II hyperlipoproteinemia.21 It remains unclear whether clinical risk factors associated with CAVD presence also predict disease progression. For example, the association between serum LDL levels and CAVD was relatively weak in the CHS study, whereas the MESA data show no association between serum LDL and CAVD in adults older than 65 years.6,18-20 Serum inflammatory markers have not been shown to be correlated with disease progression.22
| Clinical Factor | OR (95% CI) | Reference |
|---|---|---|
| Age | 2.18 (2.15-2.20) per 10-y increase | Stewart et al6 |
| 2.0 (1.7-2.3) per 10-y increment | Stritzke et al18 | |
| Male sex | 2.03 (1.7-2.5) | Stewart et al6 |
| Lp(a) | 1.23(1.14-1.32) 75th vs 25th percentile | Stewart et al6 |
| Height | 0.84 (0.75-0.93) | Stewart et al6 |
| Hypertension | 1.23 (1.1-1.4) | Stewart et al6 |
| Active smoking | 1.35 (1.1-1.7) | Stewart et al6 |
| 1.7 (1.1-2.4) | Stritzke et al18 | |
| LDL cholesterol | 1.12 (1.03 vs 1.23) 75th vs 25th percentile | Stewart et al6 |
| Total cholesterol | 1.2 (1.1-1.3) per increase of 20 mg/dL | Stritzke et al18 |
| Metabolic syndrome | 1.67 (1.21-2.31) | Katz et al19 |
| Diabetes | 2.06 (1.39-3.06) | Katz et al19 |
| Renal failure (on dialysis) | 2.47 (1.20-5.09) | Wongpraparut et al20 |
Early CAVD (aortic sclerosis) progresses to severe valve obstruction (aortic stenosis) in some, but not all, individuals. Progression from sclerosis to stenosis over a 5-year interval was observed in approximately 9% of the CHS population (all were older than 65 years).22 In a study of more than 2000 patients with aortic sclerosis, the average time progression from aortic sclerosis diagnosis to severe aortic stenosis was 8 years, with 10% developing mild stenosis, 3% developing moderate stenosis, and 3% developing severe stenosis.23 Over a longer time interval, it is unclear whether more individuals would have experienced progression.
Even in the absence of significant outflow obstruction, the presence of aortic sclerosis has been associated with adverse clinical outcomes, and this risk persists even in those who do not progress to aortic stenosis. In the CHS study, in which all participants had no known coronary artery disease, the relative risk of cardiovascular death over 5 years was 1.52 (95% CI, 1.12-2.05) compared with those with a normal aortic valve, even after correction for associated baseline clinical factors. A similar increase in risk was seen for myocardial infarction (relative risk = 1.40; 95% CI, 1.07-1.83), with a trend toward an increased risk of angina, heart failure, and stroke.24 In the LIFE hypertension study, the risk associated with aortic sclerosis was additive to the risk of underlying coronary artery disease for an adverse cardiovascular event (Fig. 76–5).25
Figure 76–5.
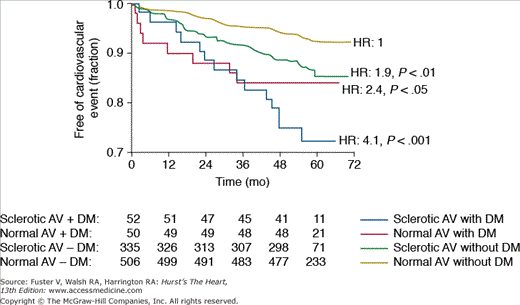
Kaplan-Meier plot showing lower freedom from cardiovascular events in patients with aortic sclerosis (green line) compared with those with normal aortic valves (AV) (yellow line). Freedom from cardiovascular events was lower in those with preexisting diabetes mellitus (DM) and was worst in those with both aortic sclerosis and DM (blue line). The number of patients at risk are at the bottom of the figure. Hazard ratios (HR) were calculated using Cox regression analysis. Reproduced with permission from Olsen et al.25
The mechanism of the increased risk associated with aortic sclerosis is unclear, but it is likely that aortic sclerosis is a marker of increased risk of atherosclerotic disease, reflecting a susceptible genetic background, an unidentified clinical risk factor,26,27 or a generalized inflammatory process.27-30 There is no evidence that the risk associated with aortic sclerosis is related directly to valve dysfunction or to progressive outflow obstruction.
A congenitally bicuspid aortic valve (BAV) is common, present in 1% to 2% of all people; 70% to 80% of cases are in men.31-33 In some cases, small family studies suggest an autosomal-dominant inheritance pattern.34 Recently, in a large family with 11 cases of congenital heart disease in 5 generations, a specific defect in the NOTCH1 gene was identified in association with a BAV and calcific aortic stenosis.35 A second NOTCH1 mutation was identified in a separate smaller family, also associated with BAV. These findings suggest a role for NOTCH1 regulation of transcription during development, as well as a role in prevention of tissue calcification.
In a small subset of patients, a BAV occurs in association with other congenital abnormalities. A BAV is present in more than 50% of patients with an aortic coarctation.36 Conversely, approximately 10% of patients with BAV also have aortic coarctation; therefore, identification of one of these findings mandates evaluation for the other. A BAV is present in approximately 10% of women with Turner syndrome and also is seen in some types of complex congenital heart disease, such as Shone syndrome (multiple left heart obstructive lesions).
The BAV phenotype may be described based on leaflet morphology. The most common phenotype, occurring in 80% of patients, is congenital fusion of the right and left coronary cusps, resulting in valve opening in a right-left orientation with larger right-sided leaflet (Fig. 76–6).37 Congenital fusion of the right and noncoronary cusps occurs in the remaining 20% of cases, resulting in an anterior-posterior opening with a larger anterior leaflet. In both phenotypes, there may be a ridge of tissue, or raphe, where the leaflets would normally separate. When the aortic valve is closed in diastole, it may be difficult to distinguish between a raphe and a closed commissure by clinical imaging; therefore, BAV diagnosis depends on visualization of the open leaflets in systole.
Figure 76–6.
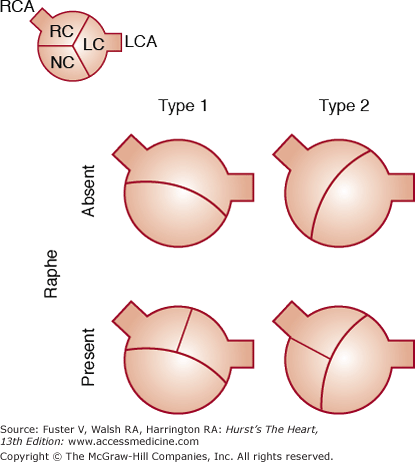
Schematic of bicuspid aortic valve (BAV) leaflet phenotypes from a parasternal short-axis view on echocardiography. Small inset top left depicts a normal trileaflet aortic valve in a similar orientation with right coronary cusp (RC), left coronary cusp (LC), noncoronary cusp (NC), and ostia of the coronary arteries. Type 1 shows congenital fusion of the right and left coronary cusps, the most common BAV phenotype, which occurs in approximately 80% of patients. The remainder of patients have the “type 2” phenotype with congenital fusion of the right and noncoronary cusps. In both types, there may be a raphe, or ridge, in the larger cusp where a commissural separation would normally occur. Reproduced with permission from Schaefer et al.37
The abnormal mechanical stress and altered shear stress of a BAV are associated with progressive valve calcification, although genetic factors, such as the NOTCH1 gene, may predispose these patients to abnormal tissue calcification.3,35 At the tissue level, calcific stenosis of a BAV appears no different from calcific stenosis of a trileaflet valve, and the term calcific aortic valve disease (CAVD) encompasses both bicuspid and trileaflet valve anatomy.
A BAV may be incompetent due to inadequate leaflet coaptation. In addition, in a subset of cases, the BAV phenotype is associated with an aortopathy (Fig. 76–7). The aortic tissue in BAV patients shows loss of elastic fibers, with smooth muscle cell disarray, increased tissue metalloproteinases, and increased apoptosis.38,39 Compared with matched patients with a trileaflet aortic valve, patients with BAV have larger ascending aortic dimensions, a faster rate of aortic dilation, and altered aortic elasticity.40-43 The pattern of aortic dilation and risk of dissection are not related to valve hemodynamics (eg, severity of stenosis or regurgitation), but preliminary studies suggest a relationship with the specific valve morphology.41 Aortic dilation is clinically significant because the risk of aortic dissection is 5 to 10 times higher in people with a BAV versus trileaflet aortic valve.44,45
Figure 76–7.
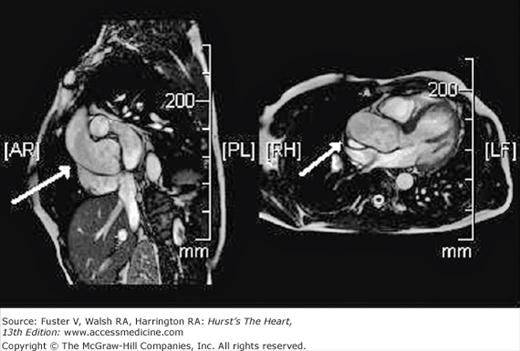
Cardiac magnetic resonance images of the aortic root in a patient with a bicuspid aortic valve. The dilated aorta (arrows) is shown as imaged from two views. With any modality of cardiac imaging, aortic dimensions in patients with a dilated aorta should be reported from several locations, including the annulus, sinus of Valsalva, sinotubular junction, ascending aorta (approximately 3-4 cm from the valve closure plane), and if available, aortic arch, descending aorta, and abdominal aorta. The aorta should be viewed from multiple angles and image planes to ensure that the maximal dimension is recorded.
Overall, long-term survival of adults with BAV does not differ from the general population; however, valve-related clinical outcomes are common. Valve function is normal in the vast majority of patients until the sixth or seventh decade of life, when CAVD changes result in severe stenosis, on average a decade earlier than stenosis of a trileaflet valve. A BAV is present in more than 50% of patients undergoing valve replacement for aortic stenosis, accounting for 66% of cases in those younger than 70 years. In patients older than 70 years, a BAV is still relatively common, present in 38% of cases.8
Although aortic stenosis is the most common sequelae of a BAV, approximately 15% to 20% of BAVs have inadequate diastolic closure leading to aortic regurgitation. Clinical presentation for significant BAV regurgitation is usually in young adulthood (20-40 years of age) and typically requires valve replacement. Indications for valve replacement are most often due to symptom onset or for prevention of heart failure based on progressive ventricular dilation or early systolic dysfunction. Rarely, clinical presentation is in childhood; these patients usually have more severe leaflet fusion or a unicuspid valve.46 Aortic complications or surgery performed primarily for aortic dilation in patients with BAV is less common than primary valve dysfunction and accounts for approximately 2% to 5% of adverse outcomes.47,48 However, compared with patients with a normal aortic valve, the risk of aortic dissection is significant. Overall, approximately 7% of aortic dissections occur in people <40 years of age. In this subgroup, 50% have Marfan syndrome, 34% have hypertensive heart disease, 19% have a familial aneurysm, 12% have had prior aortic valve replacement, and 9% have a native BAV.49
In a consecutive series of 642 adults with a BAV (mean age 35 ± 16 years, 68% men) followed for approximately 9 years, clinical outcomes occurred in 25%. These outcomes included aortic valve replacement (with or without concurrent aortic root surgery) in 22%, heart failure in 2%, aortic complications in 2%, and cardiac death in 3%. Predictors of adverse clinical outcomes were age more than 30 years and greater than mild aortic stenosis or regurgitation (Fig. 76–8).47 In a smaller study of 212 asymptomatic BAV patients followed for 20 years (mean age of 32 ± 20 years, 65% men), cardiac surgery was performed in 27%. Again, the most common outcome was aortic valve surgery in 24%, but 5% underwent isolated replacement of the ascending aorta, and 4% had surgery for aortic coarctation. Predictors of adverse outcomes in this series were older age (>50 years) and valve degeneration, defined by the degree of leaflet calcification, thickening, and reduction in mobility on echocardiography.48
Figure 76–8.
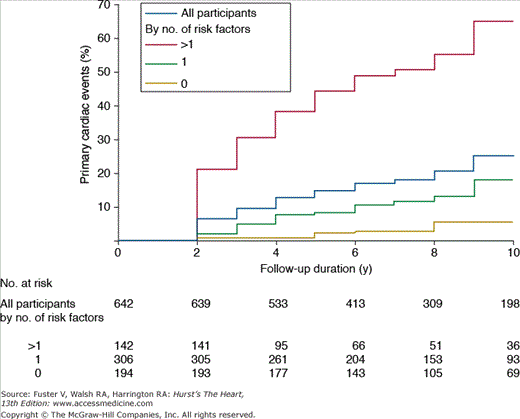
This figure shows primary cardiac events (aortic valve replacement with or without concurrent aortic surgery, heart failure, aortic complications, and cardiac death) in adults with bicuspid aortic valves followed for mean of 9 ± 5 years. Risk factors for primary cardiac events were age older than 30 years, moderate or severe aortic regurgitation, and moderate or severe aortic stenosis. The risk was highest in patients with more than one risk factor at baseline (n = 142), where primary cardiac events occurred in 65% (SD 5%). Reproduced with permission from Tzemos et al.47 Copyright © 2008, American Medical Association. All rights reserved.
Worldwide, rheumatic fever is the most common cause of valvular heart disease, with an estimated prevalence of 15.6 million persons. The geographic prevalence of deaths due to rheumatic valve disease per 100,000 population varies from as high as 6 to 7 in Southeast Asia and the Western Pacific; a mid-range of 4 to 5 in Africa, Europe, and the Eastern Mediterranean; to a low of 1.8 in the Americas.50 Rheumatic disease primarily affects the mitral valve, with secondary aortic valve involvement seen in approximately one-third of patients, typically resulting in combined aortic stenosis and regurgitation.
Rheumatic valve disease occurs as a consequence of rheumatic fever. In Europe and North America, there is an interval of approximately 15 years between acute rheumatic fever and clinically evident valve disease.51 A shorter interval between the acute illness and valve dysfunction is seen in areas with a higher prevalence of recurrent rheumatic fever. The anatomic hallmark of rheumatic valve disease is fusion of valve commissures (Fig. 76–9).52 In rheumatic valve disease, the mitral valve is uniformly affected. Commissural thickening and fusion lead to mitral stenosis. When there is chordal involvement, with thickening and shortening of the subvalvular apparatus, mitral leaflet coaptation is adversely affected and there is concomitant mitral regurgitation. In 20%-30% of cases, the aortic valve is also involved. A similar disease process affects the aortic valve with thickening of leaflet edges and commissural fusion, resulting in a central triangular systolic valve orifice and valve stenosis. With increased leaflet stiffness and deformity, aortic regurgitation may also occur. At the tissue level, rheumatic valve leaflets show thickening and fibrosis, with typical rheumatic features including Aschoff bodies, leukocyte infiltration, and prominent neovascularization. With long-standing disease, there may be superimposed calcification and fibrosis, similar to CAVD.53,54
Figure 76–9.
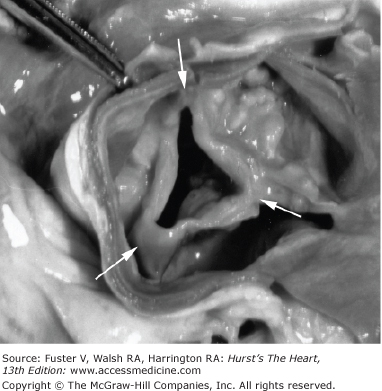
Rheumatic aortic valve disease of a trileaflet aortic valve in patient with both aortic regurgitation and stenosis. There is commissural fusion (arrows) with retraction of the leaflet tips, resulting in a triangular orifice. There is heavy calcification of the valve leaflets as well. Reproduced with permission from Otto.52
Rheumatic aortic valve disease is invariability accompanied by mitral valve involvement so that the disease course and clinical outcomes reflect multivalve disease. Mitral valve involvement is usually predominant and often defines the clinical course and management. Compared with CAVD, rheumatic aortic stenosis generally progresses more slowly, although natural history studies are limited and the disease course may be accelerated with recurrent episodes of rheumatic fever, emphasizing the importance of secondary prevention in patients with rheumatic valve disease. In addition, CAVD may become superimposed on the rheumatic valve with disease progression related to calcific disease rather than to the rheumatic process per se.
Any cause of dilation of the aortic sinuses can cause aortic regurgitation due to stretching of the valve leaflets over the enlarged orifice or to distortion of the commissures. Hypertensive and atherosclerotic diseases of the ascending aorta are often associated with a small amount of aortic regurgitation but rarely result in significant or progressive valve dysfunction. In patients with inherited connective tissue disorders, such as Marfan syndrome or familial aortic aneurysm, aortic regurgitation may vary from mild to severe, but surgery is more often primarily indicated for aortic root enlargement. Ascending aortic dissection may result in acute aortic regurgitation due to retrograde extension of the dissection into the leaflet tissue, resulting in a flail leaflet, or commissural distortion. Aortic dilation may also result from systemic inflammatory disorders, such as ankylosing spondylitis, where characteristic thickening of the aortic wall extends onto the base of the anterior mitral leaflet. Aortic regurgitation is rarely clinically significant in these patients.
Other causes of aortic valve dysfunction include tissue destruction due to endocarditis resulting in acute aortic regurgitation (see Tables 76–1 and 76–2). Iatrogenic trauma to the valve leaflets, for example, during cardiac catheterization or an electrophysiologic procedure, may cause aortic regurgitation. Blunt chest trauma, particularly deceleration injury, has been reported to cause acute valve rupture, with a postulated mechanism being an abrupt rise in aortic pressure overloading the closed leaflets in diastole. A rare cause of aortic regurgitation is rupture of a congenital valve fenestration (small discontinuities in the valve tissue common in the valve lunae, the non–load-bearing segments that normally overlap during diastole). Sudden distention of the aorta may result in transient aortic dilation with valve stretching where these areas become load bearing, with consequent leaflet tissue tearing.55
Hemodynamics
The hemodynamic consequences of aortic valve disease include obstruction to LV outflow (stenosis) and valve incompetence (regurgitation). These hemodynamic sequelae can result from any cause of aortic valve disease, and although the hemodynamic profile in an individual patient may predominantly reflect either, most patients have a combination of both lesions, to varying degrees.
The cross-sectional area of the normal aortic valve in systole is only slightly less than the size of the adjacent LV outflow tract so that blood is ejected from the LV at a low velocity (approximately 1 m/s) as a smooth column of blood with parallel streamlines across the flow area. LV and aortic pressures are nearly identical in systole with only a slight positive LV to aortic pressure difference during acceleration in early systole and a slight negative pressure difference during deceleration in late systole. There are slight changes in aortic velocity, with an increase or decrease in transaortic volume flow or cardiac output. For example, both the volume flow rate and aortic velocity are lower with LV dysfunction and higher with exercise. Over the physiologic range of changes in cardiac output, the normal aortic velocity varies from 0.8 to 2.4 m/s, regardless of body size. For any transaortic volume flow, aortic velocity and the pressure gradient increase with worsening LV outflow obstruction. The magnitude of increase in jet velocity varies with the volume flow rate. Therefore, in those with severe stenosis, but only a low stroke volume, such as in those with LV systolic dysfunction, there may only be a moderate increase in aortic velocity.
The normal adult aortic valve area is between 3.0 and 4.0 cm2. When aortic valve disease is present, the aortic velocity depends on the size of the valve orifice and transaortic volume flow. A normal cardiac output can be maintained without a significant increase in aortic velocity until valve area is reduced to approximately 25% to 30% of normal. With decreases in valve area below 1 cm2, very small changes in orifice area lead to marked changes in transvalvular pressure gradient and hemodynamic load. LV systolic pressure increases in proportion to the severity of valve obstruction, with the potential energy in the difference (or gradient) between LV and aortic pressure converted into kinetic energy as blood is ejected at high velocity across the valve (Fig. 76–10). The velocity (V) in the stenotic orifice correlates with the drop in pressure (ΔP) from the LV to the aortic, as stated in the Bernoulli equation:
ΔP = 4V2
Figure 76–10.
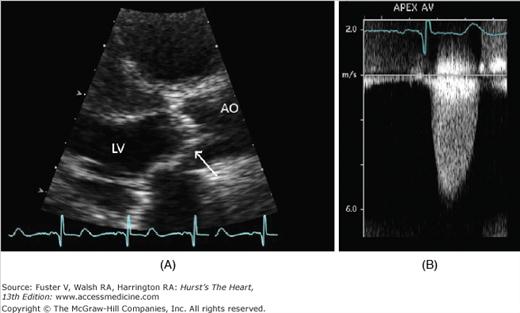
Echocardiographic images of a patients with severe aortic stenosis. A. Parasternal long-axis view of the aortic valve shows the aortic cusps of the valve are thickened and calcified with severely restricted leaflet motion during systole (arrow). B. Continuous-wave Doppler signal taken from an apical window. The peak aortic jet velocity measured 5.2 m/s.
This equation shows the relationship between instantaneous velocity and pressure measurements at any point in systole. The mean systolic pressure gradient is derived by averaging pressure gradients over the systolic ejection period.56,57
The cross-sectional area of the open aortic valve in systole is a robust measure of stenosis severity. For a given aortic valve area, the aortic velocity and pressure gradient will vary with transaortic volume flow. To account for transaortic volume flow rate, aortic valve area (AVA) can be calculated based on the continuity principle, which states that the stroke volume (SV) in the aortic valve (AV) orifice and the stroke volume just proximal to the valve in the LV outflow tract (LVOT) are equal:
SVAV = SVLVOT
Stroke volume can be measured as the cross-sectional area (CSA) of flow multiplied by the velocity integrated over the ejection period (velocity time integral, or VTI) of flow at that site:
SV = CSA × VTI
Thus the continuity equation can be solved for aortic valve area:
AVA × VTIAV = CSALVOT × VTILVOT
AVA = (CSALVOT × VTILVOT)/VTIAV
Aortic valve area also can be estimated from invasive measurement of the transaortic pressure gradient and the transaortic cardiac output using the Gorlin formula, but this approach is now rarely needed. Although valve area can be directly measured by tracing the open orifice on high-resolution images, such as transesophageal echocardiography or cardiac magnetic resonance imaging, this approach is limited by the three-dimensional anatomy of the valve, imaging artifacts due to valve calcification, and the spatial and temporal resolution of the imaging technique.58
The normal aortic valve area depends on body size, with an increase in valve area from birth to adulthood corresponding to the increase in lean body mass. In adults, valve area can be indexed for body size, typically using body-surface area, although it is not clear that indexing is necessary, nor that body surface area is the best method of indexing. Consideration of body size is most important in small adults, so that a normal (but small) valve area is not erroneously interpreted as severe valve obstruction. A simple clinical approach is to consider the ratio of the velocity in the LV outflow tract (the patient’s normal valve size) to the aortic velocity. A normal ratio is close to 1.0; a ratio of 0.25 indicates a valve area 25% of expected for that patient. The standard clinical hemodynamic parameters of aortic stenosis severity56 are as follows:
- Transaortic velocity
- Mean transaortic pressure gradient
- Aortic valve area
Definitions for mild, moderate, and severe aortic stenosis are shown in Table 76–4. This table provides only a guideline for clinical practice; in an individual patient, not all parameters will fit in the same category, and between patients, there is considerable variability in the degree of valve stenosis resulting in clinical symptoms. Other measures of aortic valve hemodynamics have been proposed and may be useful in some situations, but are not routinely recommended.59 These measures include LV stroke work loss, recovered pressure gradient, energy loss index, and valvuloarterial impedance.56
| Aortic sclerosis | Mild | Moderate | Severe | |
|---|---|---|---|---|
| Aortic jet velocity (m/s) | ≤2.5 m/s | 2.6-2.9 | 3.0-4.0 | >4.0 |
| Mean gradient (mm Hg) | — | <20(<30a) | 20-40b(30-50a) | >40b(>50a) |
| AVA (cm2) | — | >1.5 | 1.0-1.5 | <1.0 |
| Indexed AVA (cm2/m2) | >0.85 | 0.60-0.85 | <0.6 | |
| Velocity ratio | >0.50 | 0.25-0.50 | <0.25 |
Approximately 50% of adults with severe aortic stenosis have concurrent coronary artery disease. Even in the absence of associated atherosclerotic coronary disease, there is limited coronary flow reserve because the increase in coronary vessel size often fails to match the increase in LV mass. Additionally, the adequacy of myocardial oxygen delivery is limited by decreased capillary density and increased diastolic wall stress. Mismatch in myocardial oxygen supply and demand is initially evident in the subendothelium and is likely the pathophysiologic basis of angina in the absence of epicardial coronary stenosis. The severity of impaired coronary flow reserve is directly related to the severity of valve obstruction and the increase in wall stress.60-62
Aortic stenosis imposes an increased pressure load on the LV, resulting in compensatory LV hypertrophy. LV hypertrophy is defined as an increase in LV mass when compared with normal standards based on body size and sex and may be due to an increase in wall thickness with a normal chamber size (concentric hypertrophy) or an increase in LV size with relatively normal wall thickness (eccentric hypertrophy). When aortic stenosis is present, a compensatory increase in LV wall thickness serves to maintain normal wall stress (σ) because wall stress is proportional to LV pressure (P) and radius (r) and inversely related to wall thickness (Th):
σ = (P × r)/2Th
Thus the typical response of the left ventricle as aortic stenosis severity gradually progresses is symmetrically increased LV wall thickness, with no change in chamber size.63-65 Concentric hypertrophy is primarily due to additional sarcomeres aligned in parallel with a corresponding increase in cardiac myocyte size. There also may be an increase in interstitial tissue with fibrosis, which contributes to the long-term diastolic dysfunction associated with aortic valve disease.
The presence and severity of LV hypertrophy varies from patient to patient. Concurrent hypertension may result in excessive LV hypertrophy due to the additional hemodynamic afterload of increased systemic vascular resistance. Although somewhat related to the severity of valve obstruction, not all patients develop hypertrophy. An increase in LV mass is seen in 81% of women but only 54% of men with severe aortic stenosis.66 In women, the increase in LV mass is due to a marked increase in wall thickness with a small chamber size and normal or hyperdynamic systolic function, with evidence of diastolic dysfunction.66,67 In contrast, men more often have only a mild increase in wall thickness with an increase in chamber size.
LV hypertrophy is associated with diastolic dysfunction early in the disease course. By echocardiography, a pattern of impaired early diastolic relaxation is seen with a reduced early filling velocity on Doppler tracings and increased dependence on the atrial contribution to LV filling. Loss of atrial contraction with atrial fibrillation may precipitate symptoms of heart failure due to a combination of increased LV filling pressures and decreased forward cardiac output. Severe diastolic dysfunction with decreased LV compliance occurs later in the disease course. Most patients develop symptoms and undergo aortic valve replacement before this point.
In most patients with aortic stenosis, LV ejection fraction and wall stress remain normal due to LV compensatory mechanisms to increase wall thickness. However, the increase in afterload may eventually exceed the compensatory LV response, resulting in afterload mismatch and impaired LV systolic function. The concept of afterload mismatch is preservation of myocardial contractile function, but impaired systolic function due to high afterload. In this scenario, LV systolic function should improve if afterload is relieved with replacement of the stenotic valve. Clinical experience supports this concept, with LV ejection fraction usually improving after valve replacement for severe aortic stenosis (average improvement ∼10 ejection fraction units).68
A small subset of patients with aortic stenosis develop impaired myocardial contractility that does not improve after valve replacement. Most of these patients have decreased systolic function due to coronary artery disease with prior myocardial infarction or a concurrent cardiomyopathy. A few have long-standing, untreated severe aortic stenosis, resulting in irreversible myocardial dysfunction. This is a complication that can be prevented by valve replacement at symptom onset or at onset of LV dysfunction, as recommended in current guidelines.
Although aortic valve area varies less than aortic velocity (or pressure gradient), with changes in transaortic volume flow rate, the degree of valve leaflet opening can vary in some situations.69 At the extreme, there is no aortic leaflet motion when there is no transaortic flow, even with normal valve leaflets. An example of this is an LV assist device which pumps blood directly from the LV apex to the ascending aorta, bypassing the aortic valve. In patients with aortic stenosis and concurrent LV systolic dysfunction, full aortic leaflet opening may be reduced due to a low stroke volume, rather than to severe valve obstruction. In this scenario, the calculated valve area reflects the actual leaflet opening, not the maximal leaflet opening possible if cardiac output were normal. Thus it can be difficult to identify severe stenosis resulting in LV dysfunction (true stenosis) from primary myocardial dysfunction with only moderate aortic stenosis (pseudostenosis). Distinguishing these patients can be achieved with evaluation of valve hemodynamics during low-dose pharmacologic augmentation of cardiac output with either dobutamine (via increased contractility) or nitroprusside (via vasodilation).70,71 With these modalities, the change in aortic velocity and valve area is assessed as the transaortic volume flow rate increases.56 Severe stenosis is present if aortic velocity increases to at least 4.0 m/s or if aortic valve area remains 1.0 cm2 or smaller in the setting of increased stroke volume or ejection fraction by at least 20%. These patients benefit from valve replacement (Fig. 76–11).72,73 In contrast, stenosis is only moderate if aortic velocity remains less than 4.0 m/s and valve area increases to greater than 1.0 cm2 with an increase in stroke volume. For these patients, valve replacement is less likely to be beneficial. The failure of ejection fraction and transaortic flow rate to increase at all with dobutamine is called lack of contractile reserve and connotes poor prognosis with either medical or surgical therapy.74-76
Figure 76–11.
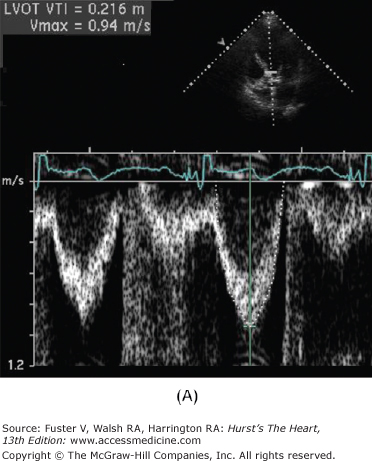
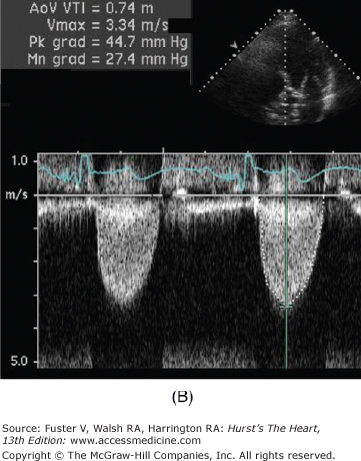
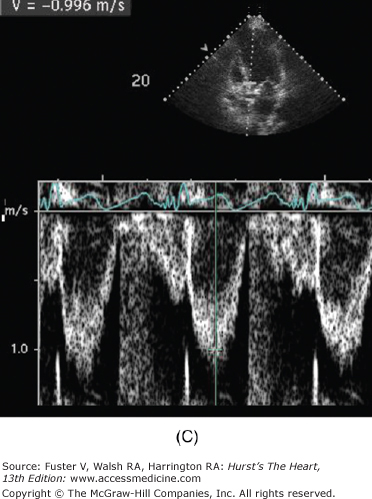
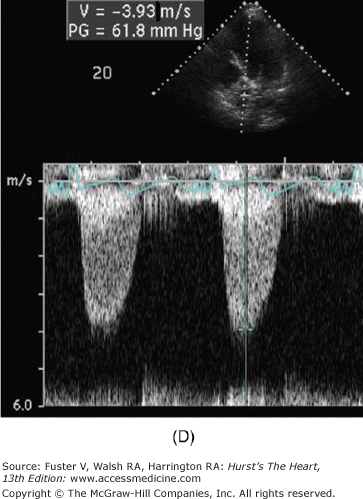
Low-dose dobutamine echocardiogram in a patient with aortic stenosis and left ventricular dysfunction (resting ejection fraction 35%). The left ventricular outflow tract (LVOT) diameter is 2.2 cm, with LVOT area of 3.8 cm2. At rest, the LVOT velocity (A) and aortic jet velocity (B) measured 0.9 m/s and 3.3 m/s, respectively (LVOT/aortic valve [AV] velocity ratio 0.27), corresponding to a peak gradient 45 mm Hg and mean gradient 27 mm Hg. With normal systolic function, this would be consistent with moderate aortic stenosis. However, with systolic dysfunction, aortic stenosis severity may be underrepresented. This is suggested by the LVOT/AV velocity ratio which is low at 0.27. At 20 μg/kg/min of dobutamine, the ejection fraction increased to 44%, the LVOT velocity measured 1.0 m/s (C), and the aortic jet velocity measured 3.9 m/s (D) (LVOT/AV velocity ratio 0.26). Despite the increase in ejection fraction, the LVOT/AV velocity ratio and the aortic valve area (1.0 cm2) were comparable between rest and 20 μg/kg/min of dobutamine (1.0 cm2). This is consistent with a fixed valve obstruction. With improved LV systolic function, valve hemodynamics is consistent with moderate to severe aortic stenosis.
Low-output aortic stenosis also may be seen in patients with a normal ejection fraction but small transaortic stroke volume.77 These patients have normal LV systolic function both visually and on measurement of ejection fraction but have a small LV chamber size due to myocardial hypertrophy, often in conjunction with diastolic dysfunction that further limits ventricular filling. Although aortic velocity and pressure gradient are relatively low in these patients, calculated aortic valve area is small and correctly reflects the severity of valve obstruction. Concurrent systemic hypertension can also hinder interpretation of aortic valve hemodynamics because increased systemic vascular resistance may reduce forward stroke volume, resulting in underestimation of the severity of valve disease. Ideally, valve stenosis severity should be remeasured after normalization of blood pressure in these patients.78,79
Incomplete closure of the aortic valve in diastole results in retrograde flow from the aorta back into the LV in diastole. The volume of retrograde blood flow is determined by the diastolic pressure difference between the aorta and LV, the size of the regurgitant orifice, the duration of diastole, and the relative compliance of the aorta and LV.80 Both a greater pressure difference between the chambers and a larger orifice area are associated with a greater degree of regurgitation. However, the interaction of physiologic variables affecting regurgitant severity can be complex. For example, with exercise, the rate of backflow per heartbeat increases due to the increase in systemic blood pressure, but the duration of diastole decreases due to tachycardia, so that regurgitant volume is unchanged.
Aortic regurgitant severity is described in terms of the amount of backflow of blood across the aortic valve in diastole.81,82 The volume leaking across the valve on each beat is multiplied by heart rate to yield regurgitant volume, measured in liters per minutes. Regurgitant fraction is the ratio of regurgitant volume to the total volume of blood ejected by the ventricle, expressed as a percentage. The actual size of the regurgitant orifice, or regurgitant orifice area (ROA), is likely the most robust measure of disease severity. This is calculated based on the continuity principle, as described for aortic stenosis. Regurgitant orifice area is calculated by dividing the regurgitant volume on a single beat (mL or cm3) by the velocity time integral of flow (VTIRJ) in the regurgitant jet through the narrowed orifice (cm):
ROA (cm2) = RV(cm3)/ VTIRJ (cm)
Regurgitant volume can be measured by Doppler echocardiography as the difference between the total stroke volume ejected by the ventricle forward across the aortic valve (in systole) and the forward stroke volume delivered to the systemic vasculature measured by the antegrade flow rate across the pulmonic or mitral valve. Total stroke volume can be measured by echocardiography, cardiac MRI, or LV angiography as the difference between end-diastolic and end-systolic volume. Regurgitant volume also can be measured by cardiac MRI based on systolic and diastolic Q-flow in the proximal ascending aorta. Continuous wave Doppler echocardiography is required to measure the velocity time integral of flow in the aortic regurgitant jet, which allows calculation of regurgitant orifice area (Table 76–5).83
| Parameter | Mild | Moderate | Severe |
|---|---|---|---|
| Jet width/LVOT | <25% | 25%-65% | >65% |
| Vena contracta (cm) | <0.3 | 0.3-0.6 | >0.6 |
| Pressure half-time (ms) | >500 | 200-500 | <200 |
| Regurgitant volume (mL/beat) | <30 | 30-60 | >60 |
| Regurgitant fraction (%) | <30 | 30-50 | >50 |
| Regurgitant orifice area (cm2) | <0.10 | 0.1-0.3 | >0.30 |
Aortic pulse pressure is increased in patients with aortic regurgitation because increased antegrade stroke volume results in high systolic aortic pressure, whereas backflow of blood across the valve in diastole results in a low diastolic pressure. The distance that retrograde diastolic flow in the aorta extends downstream correlates with regurgitant severity; moderate regurgitation is associated with holodiastolic flow reversal in the proximal descending aorta, whereas severe regurgitation results in holodiastolic flow reversal in the abdominal aorta (Fig. 76–12). With chronic regurgitation, LV diastolic pressures are usually normal because a more compliant LV allows a large LV volume at a low diastolic pressure (a shallow slope of the diastole pressure-volume relationship). This physiology is reflected in the velocity of the aortic regurgitant flow, with the velocity and timing reflecting the instantaneous aortic to LV pressure difference. With chronic regurgitation, aortic regurgitation is high velocity, reflecting a large pressure difference, with a flat slope from early to late diastole, reflecting normal LV diastolic pressures and only a slight decrease in diastolic aortic pressure (see Fig. 76–12).
Figure 76–12.
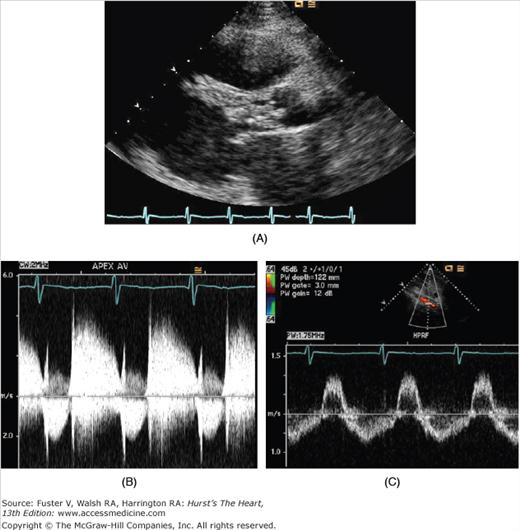
Echocardiographic images from a patient with severe, acute aortic valve endocarditis. A. There is a large vegetation that fills the left ventricular (LV) outflow tract in the parasternal long axis view. B. Continuous-wave Doppler tracing from the apex shows regurgitant flow in diastole that is as dense as antegrade flow. There is a steep deceleration slope of the aortic regurgitant jet, suggesting rapid equalization of pressures between the aorta and LV during diastole. C. In the pulse wave Doppler sample from the abdominal aorta, there is holodiastolic flow reversal in the abdominal aorta, consistent with severe aortic regurgitation.
Coronary blood flow is altered by aortic regurgitation by two factors. First, decreased diastolic aortic pressure results in a lower pressure difference between the aorta and coronary vasculature and a decreased diastolic perfusion pressure. Second, although increased LV mass and wall stress lead to a compensatory increase in coronary artery size, this increase is often inadequate to meet the higher LV oxygen demands.84-86
With chronic aortic regurgitation, the LV dilates, initially with increased compliance, to maintain normal forward stroke volume and normal aortic diastolic pressure (Fig. 76–13).80 LV volume reflects total stroke volume, which increases in direct proportion to the severity of aortic regurgitation. In addition to the LV volume overload imposed by the regurgitant lesion, there is also progressive increases in LV pressure as the larger stroke volume is ejected against peripheral systemic vascular resistance. Despite increased end-systolic pressure, wall stress is maintained by a compensatory increase in wall thickness. Combined volume and pressure overload results in a marked increase in LV mass (much greater than with any other valve lesion) and a substantial increase in LV size. The shape of the LV is affected, becoming more spherical, with apical rounding to accommodate the volume load.
Figure 76–13.
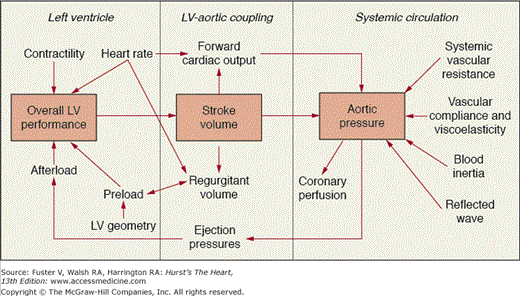
Pathophysiology of chronic aortic regurgitation. Left ventricular (LV) stroke volume includes forward cardiac output and regurgitant volume. Although increased stroke volume increases systemic systolic ejection pressure and aortic pressure, increased regurgitant volume lowers systemic diastolic pressures and, as a result, coronary perfusion. Compensatory mechanisms of the LV to maintain cardiac output despite increased regurgitant volume include dilation and remodeling of the LV. When compensatory mechanisms fail, there is adverse effect on LV contractility. The volume of regurgitant flow is determined by the diastolic pressure difference between the aorta and LV, the size of the regurgitant orifice, the duration of diastole, and the relative compliance of the aorta and LV. Modified with permission from Borow and Marcus.80
Generally, LV systolic function remains normal for many years with chronic aortic regurgitation, even as the ventricle dilates. Clinical evaluation for diastolic function is problematic as the LV accommodates both normal LV inflow across the mitral valve and the diastolic flow across the incompetent aortic valve. Chronic volume and pressure overload do eventually result in LV systolic dysfunction. Initially, systolic performance is impaired due to afterload mismatch so that LV size and systolic function return to normal if valve replacement is promptly performed. However, in more advanced cases, patients develop irreversible LV dysfunction that does not improve or only partially improves after valve replacement. Optimal timing of valve replacement in these patients is challenging because clinical measures of ventricular performance vary with changes in preload and afterload, both of which are altered in chronic aortic regurgitation.
Both prospective natural history studies and retrospective surgical series have shown that the strongest predictors of postoperative ejection fraction in patients with chronic regurgitation are the preoperative ejection fraction and end-systolic dimension (Fig. 76–14).87,88 Ejection fraction is dependent on preload and afterload so is not an ideal measure, whereas end-systolic dimension is relatively independent of preload and only mildly affected by afterload. These parameters are now used in clinical practice to guide timing of valve replacement in adults with chronic asymptomatic aortic regurgitation.
Figure 76–14.
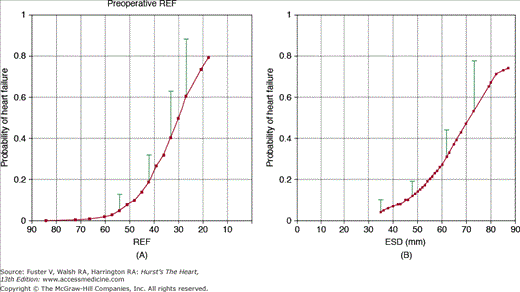
In patients with chronic aortic regurgitation who underwent aortic valve replacement, the strongest predictors of postoperative heart failure are the preoperative ejection fraction (A) and end-systolic dimension (B). With decreasing ejection fraction and increased left ventricular systolic dimension, the prevalence of postoperative heart failure increased. Reproduced with permission from Tornos et al.88
The physiology of acute aortic regurgitation differs from that of chronic aortic regurgitation. Unlike the gradual process of ventricular adaptation to an increasing volume load, in acute regurgitation, the nondilated left ventricle is suddenly exposed to acute volume overload; the left ventricle is typically small, with markedly elevated diastolic pressure.89 In the extreme case, aortic and LV end-diastolic pressure are equalized. Forward cardiac output is reduced, even when systolic function appears normal, because the total stroke volume can increase only slightly with a nondilated ventricle, and much of the blood ejected in systole returns to the LV immediately in diastole and thus does not reach the systemic vasculature. Both physical examination findings (normal pulse pressure, lack of peripheral signs of aortic regurgitation) and Doppler echocardiography (steep slope of the aortic regurgitant velocity curve) reflect this physiology.



