Aortic Valve Disease
William J. Stewart
Blase A. Carabello
Aortic Stenosis
Historical Perspective
Until about 1960, patients with aortic stenosis (AS) who developed classic symptoms of angina, syncope, or congestive heart failure (CHF) had only a few years to live, and survival was unaffected by medical therapy. Now the age-corrected survival after successful aortic valve replacement (AVR) mimics the normal population (1). Excellent outcome is predicated on proper timing of reliably safe surgery, and use of dependable “substitute” valves with excellent hemodynamic profiles. AS is primarily a disease of aging, and its incidence is increasing in our population, occurring in approximately 3,000,000 Americans.
Diagnosis of aortic valve disease has also undergone substantial refinement. The physician’s bedside assessment of the severity of stenosis remains the first stage of diagnosis. Doppler echo provides accurate noninvasive measures of the transvalvular gradient (2,3), based on valvular velocity and the modified Bernoulli equation, and the aortic valve area (AVA) (4), based on the continuity equation. Left ventricular (LV) catheterization using the retrograde approach provides direct measurement of gradients and estimation of AVA.
After the development of cardiopulmonary bypass in 1953, Harken et al. (5) were first to perform successful AVR with a subcoronary mechanical prosthesis in 1960. Ross pioneered the aortic valve homograft in 1962 (6), and in 1967 transfer of the pulmonary valve to the aortic position (pulmonary autograft) (7), an approach that gained appreciable acceptance in the 1980s and 1990s before declining in popularity more recently.
Many aortic valve prostheses have been used and discarded over the years. The Starr-Edwards ball-and-cage prosthesis remained popular for most of the last 30 years, declining recently with the advent of lower profile mechanical valves. Carpentier et al. (8) developed a glutaraldehyde-preserved porcine heterograft in 1969. The bovine pericardial heterograft, after the first series developed early failure (9), was improved and is among the most widely used today (10). Tissue valves available today include stent-mounted pericardial valves, stent-mounted porcine valves (now declining in usage), stentless porcine valves, cryopreserved homografts (allografts), and pulmonary autografts.
Etiology
An Inflammatory Process Similar to Atherosclerosis
Several etiologies of valvular AS have been described, including senile calcific, bicuspid, rheumatic, and congenital varieties. In developed countries, the most common cause of adult-acquired AS at present is a chronic inflammatory and fibrotic process of the aortic valve very similar to arterial atherosclerosis. The progressive fibrosis and calcification (11) can occur on a bileaflet or a trileaflet valve, it just occurs earlier in life in the bicuspid valve.
About one percent of Americans are born with a bicuspid aortic valve. In most of these, the valve opens well at birth, but gradual and progressive fibrosis leads to stenosis in approximately one third (12,13), presumably because of flow characteristics of the bicuspid architecture increase chronic subclinical injury of the valve tissue and accelerate the atherogenic
process. Because bicuspid aortic valves occur more frequently in men, AS also has a male predominance. When it occurs, clinically significant stenosis of a bicuspid valve typically develops in the fifth or sixth decades of life, although it may occur earlier or later. In contrast, rheumatic AS, on average, becomes symptomatic in the fourth decade, and congenital AS becomes symptomatic in the first to third decades (Fig. 23.1) (14). Although the exact incidence of AS in patients born with tricuspid aortic valves is unknown, it is probably less than 1%, but is increasing with our aging population. In patients with a previously normal tricuspid aortic valve who eventually develop AS, the same atherosclerosis-like process, involving inflammation and calcification, leads to symptomatic stenosis, occurring typically in the seventh and eighth decades (14).
process. Because bicuspid aortic valves occur more frequently in men, AS also has a male predominance. When it occurs, clinically significant stenosis of a bicuspid valve typically develops in the fifth or sixth decades of life, although it may occur earlier or later. In contrast, rheumatic AS, on average, becomes symptomatic in the fourth decade, and congenital AS becomes symptomatic in the first to third decades (Fig. 23.1) (14). Although the exact incidence of AS in patients born with tricuspid aortic valves is unknown, it is probably less than 1%, but is increasing with our aging population. In patients with a previously normal tricuspid aortic valve who eventually develop AS, the same atherosclerosis-like process, involving inflammation and calcification, leads to symptomatic stenosis, occurring typically in the seventh and eighth decades (14).
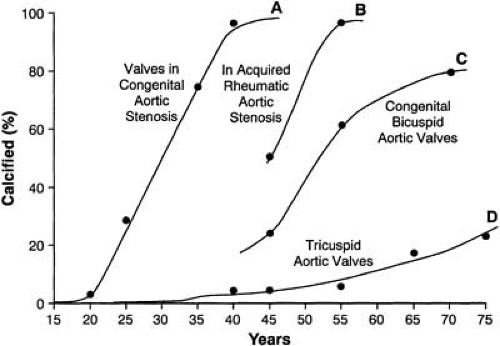 FIGURE 23.1. Timing of onset of symptoms and significantly calcified and stenotic valves versus the major etiologies of AS, including (A) congenital, (B) bicuspid, (C) rheumatic, and (D) senile. (Source: From Campbell M. Calcific aortic stenosis and congenital bicuspid aortic valves. Br Heart J 1968;30:606–616 , with permission.) |
Otto et al. (15) have noted that the early lesion of AS is characterized by subendothelial thickening on the aortic side of the leaflet. Thickening occurs as the elastic lamina is displaced by the accumulation of cellular lipid infiltration and extracellular mineralization. There is proliferation of smooth muscle cells and lipid-laden foam cells resembling the plaque of coronary atherosclerosis (12).
Standard risk factors for coronary atherosclerosis are associated with acceleration of the thickening of the aortic valve. In fact, even mild degrees of fibrosis, termed aortic sclerosis, is a manifestation of an atherosclerotic process on the aortic valve that eventually leads to AS (16,17,18,19). Aortic sclerosis itself is associated with a significantly worse prognosis, possibly also owing to its link to atherosclerosis in all parts of the cardiovascular system (19).
Cholesterol, lipoprotein disorders, smoking, male gender, and age may accelerate the fibrotic effects (20) and development of AS. In patients with familial hypercholesterolemia, duration of exposure to high cholesterol concentrations correlates with development of AS (21). Acquired AS involves the same atherosclerosis-like histology and pathophysiology, and the same ultimate outcome, in bicuspid and tricuspid valves, only at an earlier age in the former.
Rheumatic Valvular Disease
In developed countries, rheumatic fever is the cause of AS in a minority of patients, accounting for less than 20% of the total (11). When rheumatic fever is the cause of AS, the mitral valve is also affected in most patients. Thus, the diagnosis of rheumatic AS should probably not be made in the presence of a completely normal-appearing mitral valve by echocardiography unless a diagnosis of previous rheumatic fever is ironclad (13).
Radiation-Induced Aortic Stenosis
There is an increased prevalence of AS following mediastinal radiation occurring two decades or more after therapy, sometimes associated also with radiation-induced disease of the coronary arteries, pericardium, myocardium, lungs, esophagus, or thyroid gland (22).
Congenital Aortic Stenosis
Congenital LV outflow tract obstruction occurs at valvular, subvalvular, and supravalvular levels. Williams syndrome is the association of supravalvular AS with elfin facies and hypercalcemia. It is sometimes associated with peripheral pulmonary arterial stenosis, which tends to improve with time, whereas the supravalvular AS tends to progress with time, probably owing to failure of growth of the sinotubular junction (23).
Subvalvular AS can be caused by a membranous ring or by a fibromuscular tunnel. In addition, some patients are born with small aortic roots, representing a mild form of aortic atresia (see Chapter 30). Congenital valvular AS is usually caused by cusp fusion of bicuspid, or unicuspid valves, and may present with severe heart failure in infancy. Congenital valvular AS differs greatly from adult-acquired AS, with more common incidence of normal or supernormal ejection performance (24,25), often with extensive hypertrophy, and more frequent sudden cardiac death (26). Thus, in pediatric patients, valve replacement should be recommended if the mean transvalvular gradient exceeds 50 mm Hg, even in the absence of symptoms. Because the commissural fusions are not densely calcified in congenital valvular AS, balloon aortic valvotomy is a reasonable option in asymptomatic patients with noncalcified congenital AS whose mean transvalvular gradient exceeds 50 mm Hg (27).
Clinical Presentation
Natural History
The natural history of acquired AS was depicted by Ross and Braunwald (Fig. 23.2) and confirmed by recent studies (28,29,30,31). The patient who eventually develops severe AS remains asymptomatic with a nearly normal survival for a long latency period. During this time, the valve slowly
develops fibrosis, inflammation, and calcification, but the patient remains well. However, once the symptoms of angina, syncope, or CHF develop, survival is dramatically shortened. Approximately 35% of patients who become symptomatic present with angina (32). In this group, 50% survive for only 5 years unless AVR is performed. In the 15% of patients who present with syncope, 50% survive for only 3 years; for the 50% with AS who present with symptoms of CHF, mean survival is less than 2 years unless the aortic valve is replaced. The risk of sudden cardiac death in asymptomatic or minimally symptomatic patients is rare, about 0.5% per year, whereas it increases to 2% per month after the development of symptoms (30,31). If the risk of death without surgery in a few years is less than that associated with AVR with available therapies, it seems reasonable to wait until the development of symptoms. However, declining surgical mortality rates have changed this, with earlier operation becoming more common (see section on AVR in asymptomatic AS).
develops fibrosis, inflammation, and calcification, but the patient remains well. However, once the symptoms of angina, syncope, or CHF develop, survival is dramatically shortened. Approximately 35% of patients who become symptomatic present with angina (32). In this group, 50% survive for only 5 years unless AVR is performed. In the 15% of patients who present with syncope, 50% survive for only 3 years; for the 50% with AS who present with symptoms of CHF, mean survival is less than 2 years unless the aortic valve is replaced. The risk of sudden cardiac death in asymptomatic or minimally symptomatic patients is rare, about 0.5% per year, whereas it increases to 2% per month after the development of symptoms (30,31). If the risk of death without surgery in a few years is less than that associated with AVR with available therapies, it seems reasonable to wait until the development of symptoms. However, declining surgical mortality rates have changed this, with earlier operation becoming more common (see section on AVR in asymptomatic AS).
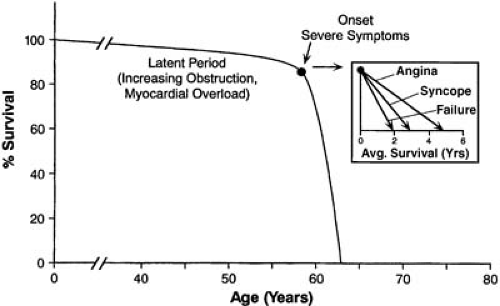 FIGURE 23.2. The natural history of AS. A long latent period with a nearly normal survival rate is present until the symptoms of angina, syncope, or CHF develop. At that time, a precipitous decline in survival occurs unless the problem is surgically corrected. Inset: The prognosis is worst with presenting symptoms of CHF, second worst with syncope. (Source: From Ross J Jr, Braunwald E. Aortic stenosis. Circulation 1968;38[Suppl 5]:V–61 , with permission.) |
The progression of AS is more rapid, and prognosis without AVR is worse, in patients who have moderate or severe valvular calcification and also in those who have a rapid increase in their Doppler-derived gradient (33,34,35). Similarly, smoking, hypercholesterolemia, and elevated serum creatinine and calcium levels have been associated with a more rapid rate of progression of AS (36).
Pathophysiology of Aortic Stenosis and Its Relation to Symptoms
As AS worsens and the gradient increases, a progressive pressure overload is placed on the left ventricle. Increased systolic wall stress signals development of concentric LV hypertrophy (LVH) (37), accompanied by a dissociated and complex modulation of various genes responsible for contraction and relaxation. LVH also involves the renin–angiotensin system (38,39), and other pathways involving LV remodeling (40) associated with altered myocardial matrix metalloproteinase activity and expression. Increased muscle mass allows the ventricle to generate increased pressure while maintaining normal wall stress to compensate for the pressure load. The Laplace equation for wall stress is used to approximate afterload on a given area of myocardium.
As pressure in the numerator increases, increased thickness in the denominator offsets the pressure overload, and wall stress remains normal. However, as the pressure overload progresses, the hypertrophy that develops becomes involved in the pathogenesis of symptoms (e.g., subendocardial ischemia and arrhythmias), and thus a worsened outcome.
Angina
Angina, representing myocardial ischemia, occurs in AS because myocardial oxygen supply decreases and is outstripped by demand, which increases (41). With concentric hypertrophy, coronary flow reserve (during stress) becomes reduced from its normal five- to eightfold increase above baseline to a two- to threefold increase (Fig. 23.3) (42). Reduced flow reserve results from the endocardial compression by increased diastolic pressure, possibly with inadequate capillary ingrowth (43,44). In addition, outflow obstruction reduces diastolic perfusion time (45,46). Myocardial oxygen demand is increased in AS by increased systolic pressure and wall stress (47). Although LVH initially helps to normalize wall stress in AS, it becomes inadequate during exercise and later in the disease (48).
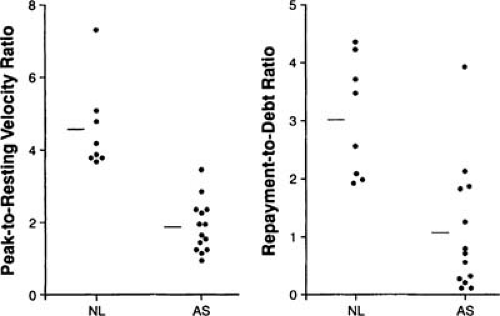 FIGURE 23.3. Shown here are ratios of peak-to-resting velocity and of repayment to debt, demonstrating the coronary reactive hyperemia responses (after a 20-second occlusion of the left anterior descending coronary artery in control patients [NL] and in patients with AS). (Source: From Marcus ML, Doty DB, Hiratzka LF, et al. Decreased coronary reserve: a mechanism for angina pectoris in patients with aortic stenosis and normal coronary arteries. N Engl J Med 1982;307:1362–1367 , with permission.) |
Syncope
Syncope in patients with AS, almost always precipitated by exertion, may be caused by an inability to increase in cardiac output during exercise, coupled with the normal decrease in total peripheral resistance. Alternatively increased intraventricular pressure during exercise may trigger a vasodepressor response (32,49,50). Supraventricular or ventricular arrhythmias may precipitate hypotension and syncope.
Congestive Heart Failure
Both systolic and diastolic myocardial dysfunction may occur in AS. This is detected by physical exam, chest radiography, and elevation of brain natriuretic peptide (BNP). The concentric hypertrophy requires a higher diastolic pressure for filling. Despite increased overall chamber stiffness because of LVH, the stiffness of each unit of myocardial volume remains normal (51) initially. However, as the disease progresses, increased collagen deposition reduces muscle compliance causing impaired LV filling with intrinsically stiffer myocardium (51,52).
Ventricular shortening depends on both afterload and contractility. Of patients with AS, 80% have excess afterload with increased peak systolic stress (Fig. 23.4) (53). The increase in pressure, the numerator of the Laplace equation, has not been normalized because of an inadequate increase in wall thickness in the denominator. Increased afterload reduces ejection performance and cardiac output and eventually decreases contractile function. Why the degree of hypertrophic compensation varies from patient to patient is only partially understood (54). Perhaps there is an inherent variability in the set point for pressure regulation of cardiac growth (55). The etiology of the decrease in contractility may include impaired calcium handling and subendocardial ischemia (56,57,58). Densification of microtubules comprising the myocardial cytoskeleton occurs in the context of inadequate hypertrophy and persistently increased systolic wall stress (59,60).
In addition to imaging studies that assess systolic dysfunction or ejection fraction (EF) in standard ways, diastolic dysfunction assessed using Doppler echocardiography, correlates with prognosis (61). The “Tei index,” which calculates isovolumic contraction time plus isovolumic relaxation time divided by LV ejection time, helps to differentiate between symptomatic AS patients with depressed and less symptomatic AS patients with preserved systolic LV function (62).
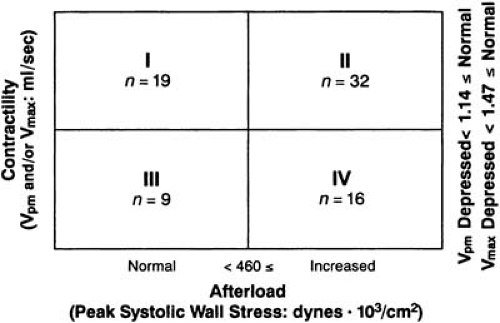 FIGURE 23.4. Here, contractility assessed as maximum dP/dT/P (Vpm and/or Vmax) is plotted against afterload assessed as peak systolic wall stress in 76 patients with AS. Of the 57 patients with some abnormality, 48 (85%) had increased afterload. (Source: From Huber D, Grimm J, Koch R, Krayenbuehl HP. Determinants of ejection performance in aortic stenosis. Circulation 1981;64:126 , with permission.) |
Gastrointestinal Bleeding
Patients with AS have an increased risk of gastrointestinal bleeding (63,64), termed Heyde syndrome. Angiodysplasia, or arteriovenous malformations of the intestines, can occur, although they are difficult to find by standard methods (63). When unexplained anemia or occult blood in stool is present, an operative strategy that requires postoperative anticoagulation seems ill advised (64).
Hemodynamics
The best measurements for assessing the severity of AS are the mean systolic gradient and AVA, which are obtained accurately both by catheterization and Doppler methods, which are both derived from similar principles. The Gorlin formula uses direct pressure measurements from a catheter and pressure manometer. The continuity equation uses direct velocity measurements from Doppler echocardiographic instruments (4). Both methods derive AVA from Torricelli’s equation:
where F is flow, A is orifice area, and V is velocity. Using Doppler methods, velocity is measured directly by determining the amount of shift in the frequency of reflected ultrasound, both in the valve orifice (by continuous wave Doppler) and in the LV outflow tract (by pulsed Doppler). The area in the LV outflow tract is calculated by measuring its diameter directly from a two-dimensional echo image and assuming circular symmetry. Outflow tract area multiplied by outflow tract velocity are divided by valvular velocity to obtain the valve area using the continuity equation.
Using catheter methods, cardiac output is measured using the Fick or indicator dilution (thermodilution) technique. Pressure gradient is measured and converted to velocity:
where g is acceleration owing to gravity and h is mean gradient. These are combined in the Gorlin formula for calculating AVA (65), which is the following:
where Q is cardiac output and h is the mean valve gradient. An alternative, third method for determining AVA is by careful planimetry of the systolic short-axis views of the aortic valve on a transesophageal echocardiography (TEE) (66), although this is not generally as accurate as the continuity equation. In some patients, TEE from deep transgastric views can record velocities representative of the transvalvular pressure gradients, although gradients derived in this way may miss the highest ones present.
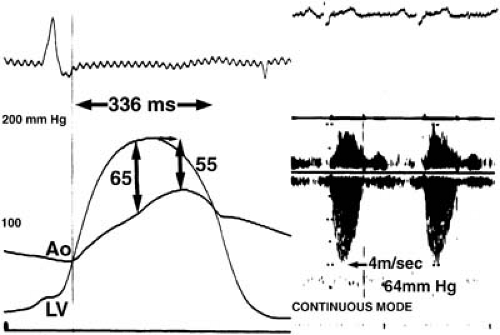 FIGURE 23.5. Micromanometer recordings of LV and ascending aortic pressure tracing (left) and continuous-wave Doppler velocity envelope (right) demonstrate correspondence of Doppler-derived peak instantaneous gradient (64 mm Hg) with peak instantaneous gradient determined by catheter measurements (65 mm Hg), which differs from the peak-to-peak gradient (55 mm Hg). The catheter- and Doppler-derived mean gradients matched perfectly (42 mm Hg). (Source: From Assey ME, Usher BW, Carabello BA, Spann JF Jr. The patient with valvular heart disease. In: Pepine CJ, Hill JA, Lambert CR, eds. Diagnostic and therapeutic cardiac catheterization, 2nd ed. Baltimore: Williams & Wilkins, 1994:692 , with permission.) |
To record pressure gradients invasively, a catheter is used to record pressure on both sides of the valve, ideally with a simultaneous recording, from which the average pressure difference during the systolic interval is measured. For Doppler methods, valvular velocity recorded by continuous wave Doppler is converted to gradient (G) using the simplified Bernoulli equation (Fig. 23.5) (2,3,67).
Both Doppler and catheter methods can accurately derive the mean systolic gradient, averaged over the systolic interval. Peak instantaneous gradient measured by Doppler has no convenient direct-pressure measurement counterpart, and specifically is not the same as peak-to-peak gradient.
Progression of Aortic Stenosis
As shown in Table 23.1, reduction in aortic orifice area from its normal 3.0 to 4.0 cm2 to approximately 1.5 cm2 results in only a small gradient. However, after that degree of stenosis has developed, small additional reductions in the orifice area lead to progressively more dramatic increases in gradient. The average increase in gradient is 7 to 10 mm Hg per year (68) but with a large and unpredictable individual variation, as much as 25 mm Hg in 1 year in occasional patients (69).
A rapid rate of progression of stenosis, as reflected by an increase in aortic-jet maximum velocity of more than 0.3 m/second in the course of a year, occurs more often in patients who have clinical cardiac events (33).
TABLE 23.1 Mean Systolic Pressure Gradient and Orifice Area in Aortic Stenosis | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


