Histology
Etiologies
Pulmonary Capillaritis
Granulomatosis with polyangitis
Microscopic polyangitis
Isolated pulmonary capillaritis
Systemic lupus erythematosus
Primary antiphospholipid antibody syndrome
Other collagen vascular disorders/connective tissue diseases
Henoch-Schönlein purpura
Behçet Syndrome
Goodpasture syndrome
Acute lung transplant rejection
Hematopoietic stem cell transplantation
Cryoglobulinemia
Drugs and medications (e.g. propylthiouracil)
Bland Pulmonary Hemorrhage
Idiopathic pulmonary hemosiderosis
Goodpasture syndrome
Systemic lupus erythematosus
Coagulation disorders
Inhalational exposures (e.g. trimellitic anhydride, isocyanates)
Drugs and medications (e.g. penicillamine, amiodarone, nitrofurantoin)
Mitral stenosis/valvular heart disease
Left ventricular dysfunction
Obstructive sleep apnea
Pulmonary veno-occlusive disease
Diffuse Alveolar Damage
Acute respiratory distress syndrome
Acute idiopathic pneumonia
Hematopoietic stem cell transplantation
Drugs and medications (e.g. cocaine inhalation)
Acute exacerbation of interstitial lung disease
Miscellaneous
Lymphangioleiomyomatosis
Human immunodeficiency virus infection
Pulmonary capillary hemoangiomatosis
Clinical Vignettes
Case One
A 19 year old man presented to the emergency room complaining of 1 week of progressive dyspnea on exertion and non-productive cough, initially thought to be a respiratory infection. On further history, he revealed a 1 month history of a non-pruritic rash on his legs and ankles. He denied any fever, chills, chest pain, sputum production, recent inhalational injury, cocaine or other drug use, or any human immunodeficiency virus (HIV) risk factors. Review of systems was positive for fatigue, malaise, abdominal pain that was worse after meals, and diffuse arthralgias, particularly of the large joints. He also endorsed multiple episodes of hematochezia (passing bright red blood per rectum) over the past month. He denied any sinus disease, gross hematuria, focal weakness, or paresthesias. His only medications were non-steroid anti-inflammatory agents on an as needed basis.
Physical exam revealed tachycardia, tachypnea, increased respiratory effort with accessory muscle use and significant hypoxemia with an oxygen saturation of 92 % while on high flow oxygen through a non-rebreather mask. He was anxious and speaking only in short sentences. His pulmonary exam revealed diffuse bilateral crackles. His abdominal examination revealed diffuse tenderness and a positive guaiac test for occult blood. His skin exam was notable for irregular, palpable, slightly raised, purpuric lesions with surrounding petechiae on his lower extremities.
The patient’s respiratory status deteriorated over the ensuing 4–5 h, ultimately requiring intubation and mechanical ventilation. Laboratory testing was notable for an elevated white blood cell count of 17,000 cells/mm3 and an elevated erythrocyte sedimentation rate of 87 mm. His laboratory testing also indicated acute renal failure with a creatinine of 1.8 mg/dl, and his urinalysis revealed both granular casts and microscopic hematuria, but no red blood cell casts. Chest imaging revealed patchy, heterogenous, diffuse bilateral infiltrates and bronchoscopy revealed an increasingly bloody return on serial aliquots. Skin biopsy confirmed a leukocytoclastic vasculitis and IgA positive immunofluorescence. The patient was diagnosed with Henoch-Schönlein purpura.
The patient was treated aggressively with intravenous corticosteroids, cyclophosphamide and plasmapharesis. He had resolution of his respiratory failure and was liberated from mechanical ventilation on hospital day #5. His renal function also subsequently returned to normal, and he was discharged to home on hospital day #16.
Case Two
A 28 year old man presented to clinic for progressive dyspnea and fatigue. The patient has a complex past medical history notable for multiple episodes of deep venous thrombosis and a known diagnosis of anti-phospholipid antibody syndrome. Further work-up for systemic lupus erythematosus and other collagen vascular diseases was negative. The patient has been maintained on chronic oral anti-coagulation for the past 4–5 years.
Approximately, 1 year ago the patient had a “flare” of his disease that began with a non-productive cough, fatigue and dyspnea, similar to his current presentation. However, with the earlier episode, he went on to develop hemoptysis and respiratory distress. Surgical lung biopsy at an outside hospital revealed alveolar hemorrhage and an underlying fibrotic non-specific interstitial pneumonitis. He was treated with intravenous corticosteroids and improved. Since that time, his oral corticosteroids have slowly been weaned, and at the time of the current presentation, he was down to 10 mg of oral Prednisone every other day. Of note, the patient also reported that he had recently resumed smoking 1/4–1/2 pack of cigarettes per day.
Physical examination was notable for a mildly elevated heart rate of 100 beats per minute and a mildly elevated respiratory rate of 20 breaths per minute. Auscultation revealed crackles at the right base, but breathing was otherwise easy, symmetric and unlabored. Pulmonary function testing revealed a forced vital capacity that was 65 % predicted and FEV1 that was 70 % predicted, but a normal diffusing capacity of carbon monoxide (DLCO) at 90 % predicted that corrected to 108 % predicted when adjusted for alveolar volume. High resolution computed tomography (HRCT) of the chest demonstrated patchy ground glass opacities (Fig. 10.1a, b). Bronchoscopy revealed diffuse alveolar hemorrhage on lavage.
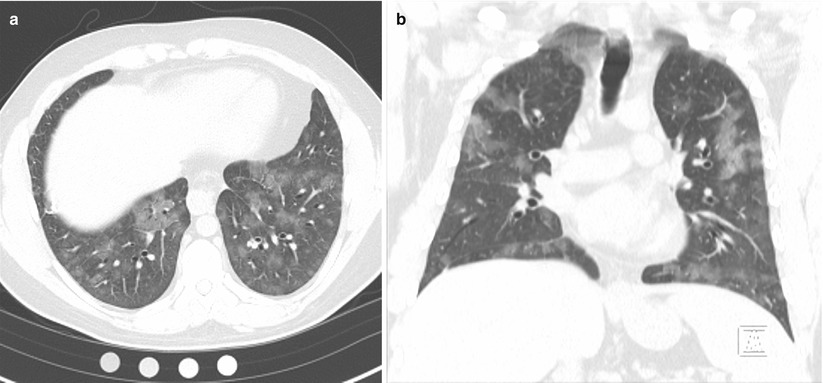
Fig. 10.1
(a, b) High resolution computed tomography images demonstrating patchy ground glass opacities consistent with alveolar hemorrhage
The patient was diagnosed with DAH secondary to recurrent anti-phospholipid antibody syndrome and was successfully treated with increased doses of oral corticosteroids and the addition of a steroid-sparing, cytotoxic agent. Upon achieving a goal maintenance dose of cytotoxic agent, the corticosteroids were successfully tapered to 5 mg of oral Prednisone daily.
Case Three
The patient is a 75 year old gentleman who was in good health and quite active until 6–8 months prior to presentation. At that time, he was noted to develop dyspnea on exertion by family members and was encouraged to seek medical attention. Pulmonary evaluation revealed significant functional impairment, and HRCT demonstrated a basilar predominant, reticular pattern of interstitial lung disease. Autoimmune serologies including anti-nuclear antibodies, anti-neutrophil cytoplasmic antibodies, rheumatoid factor, anti-Scl-70, anti-SS-A and anti-SS-B antibodies were all negative. Surgical lung biopsy revealed usual interstitial pneumonitis, and the patient was diagnosed with idiopathic pulmonary fibrosis.
Over the first few months following the diagnosis, the patient noticed a slow, steady decline in function, but over the 2–3 weeks prior to presentation, he became dramatically worse with markedly increased oxygen requirements and dyspnea that occurred with ambulating room to room. Upon presenting to clinic, he was found to be in respiratory distress with a respiratory rate of 32 breaths per minute, accessory muscle use and increased work of breathing. The patient was admitted to the Intensive Care Unit.
Further evaluation included HRCT of the chest which revealed diffuse ground glass infiltrates superimposed on an underlying fibrosing interstitial pneumonia consistent with his known diagnosis of idiopathic pulmonary fibrosis (IPF) (Fig. 10.2a, b). White blood cell count was normal, but his hematocrit was reduced at 35 %. Bronchoscopy reveal diffuse alveolar hemorrhage (Fig. 10.2c). Differential cell counts from the BAL revealed a 60 % neutrophilia, but no infectious organisms were isolated. Echocardiography confirmed normal left ventricular function and filling pressures and was otherwise unremarkable. No evidence of pulmonary embolus or other precipitant of respiratory decline could be identified. Given that no specific precipitant for the patient’s acute respiratory decline could be identified and that infection, heart failure, thromboembolic disease and other potential causes of acute lung injury were all excluded, the patient was diagnosed with an acute-exacerbation of IPF. Furthermore, the bronchoscopic finding of alveolar hemorrhage did not prove to represent clinically-significant hemorrhage and repeat serologies, ANCA testing, anti-basement membrane antibodies were all negative. Following a prolonged ICU course, he died of his respiratory failure.
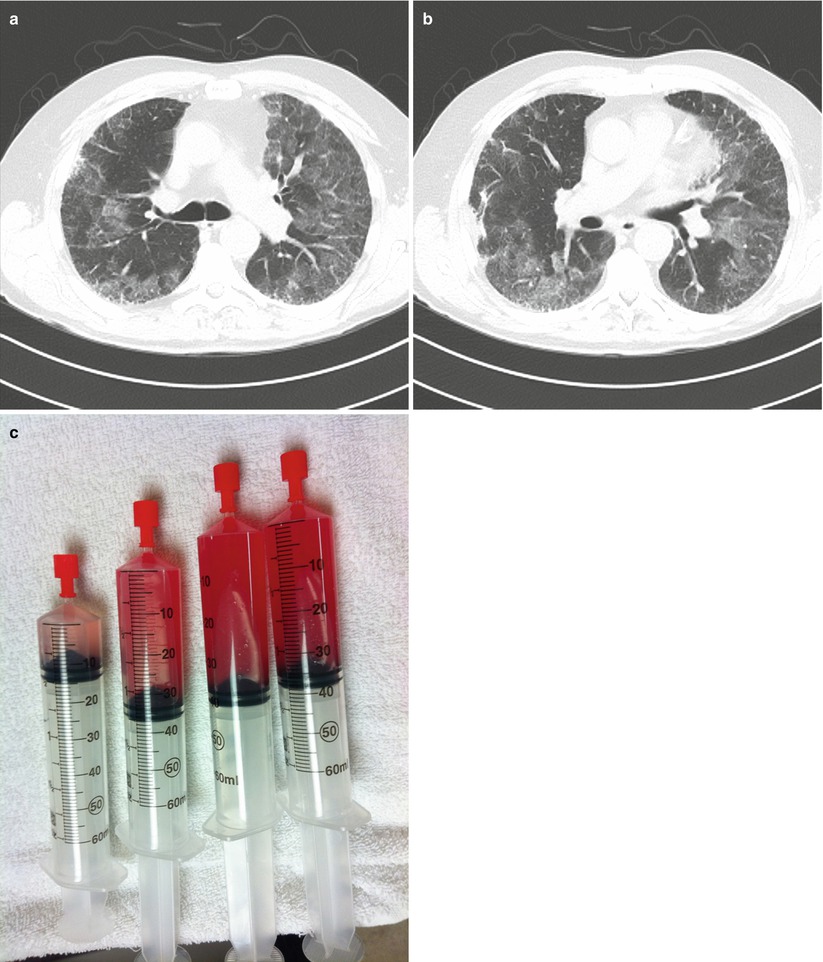
Fig. 10.2
(a, b)High resolution computed tomography images demonstrating patchy ground glass opacities superimposed on peripheral-predominant reticular infiltrates and early honeycomb changes. (c), Serial aliquots of bronchoalveolar lavage fluid demonstrating an increasingly hemorrhagic return diagnostic of alveolar hemorrhage
Clinical Presentation
Patients who present with DAH can present at any age. Patients may have a known predisposing condition such as a systemic vasculitis, collagen vascular disease, or mitral stenosis, or the DAH may represent the initial manifestation of their disease state. DAH may occur as an isolated event or with repeated episodes of bleeding. Hemoptysis, the most characteristic sign of DAH, may evolve slowly over a period of weeks (i.e. anti-phospholipid antibody syndrome [1]) or more dramatically over a period of days or hours (i.e. crack cocaine inhalation [2]). However, it has also been reported that up to one-third of cases of DAH will present without evidence of hemoptysis [3]. Additional pulmonary symptoms may include dyspnea, non-productive cough, exercise intolerance, and/or vague chest discomfort or heaviness. As mentioned, patients may present earlier in a disease course with more mild symptoms of dyspnea or hemoptysis, or they may present with fulminant disease including profound hypoxemia or respiratory failure. Constitutional symptoms may also commonly be seen including fatigue, malaise, anorexia, fever, and myalgias.
In evaluating any patient with DAH, a comprehensive and detailed history is very important. Areas to consider include: (1) Does the patient have any elements to suggest a systemic autoimmune or collagen vascular disorder? The identification of extra-pulmonary signs and symptoms may be helpful in revealing a potential underlying etiology for the DAH. For example, the identification of skin lesions consistent with a cutaneous leukocytoclastic vasculitis, the presence of destructive upper airway lesions, or the finding of inflammatory ocular disease may point the clinician towards the diagnosis of a primary small vessel vasculitis. Similarly, does the patient have a malar rash or synovitis to suggest possible systemic lupus erythematosus? (2) Does the patient have any underlying cardiac disease? Specifically, does the patient have valvular heart disease (i.e. mitral stenosis or rheumatic heart disease) or disease that might result in elevated left-sided filling pressures? (3) Does the patient take any potentially causal medications such as penicillamine or propythiouracil? Or engage in illicit drug use, such as crack cocaine? (4) Does the patient have a coagulation disorder or take any anti-coagulants that might contribute to hemorrhage?
Physical examination findings in DAH are nonspecific. Objective findings may include fever, tachypnea, tachycardia, hypoxemia, diffuse crackles/rales, bronchial breath sounds or other findings consistent with alveolar consolidation on chest auscultation. The search for extra-pulmonary findings though may be extremely fruitful as regards identifying an inciting underlying systemic disease. Such findings may include palpable purpura, conjunctivitis, septal perforation, iridocyclitis, synovitis, or focal neurologic deficits/mononeuritis multiplex.
On laboratory testing patients will be noted to have a low and/or falling hemoglobin. However, the presence of a normochromic, normocytic anemia in acutely-ill patients tends to be a non-specific finding. In the case of subclinical bleeding or recurrent bouts of DAH, iron deficiency anemia may develop as well. Generally speaking, elevations of the white blood cell counts and platelets will be noted, although thrombocytopenia may be seen in conjunction with DAH in entities such as idiopathic thrombocytopenic purpura, thrombotic thrombocytopenia purpura, hemolytic uremic syndrome, or disseminated intravascular coagulation [4, 5]. Of note, these conditions are generally associated with bland pulmonary hemorrhage rather than a capillaritis lesion. Additionally, the presence of thrombocytopenia with DAH should also raise suspicion for possible systemic lupus erythematosus (SLE) [6] or primary anti-phospholipid antibody syndrome (APLAS) [7].
Coagulation studies are critical to excluding coagulopathy as the inciting etiology of DAH (bland hemorrhage). Elevated inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein are commonly elevated, but are non-specific findings. Serologic testing for specific autoimmune disorders and immune complex mediated diseases is a necessary part of the evaluation of DAH and extremely helpful when positive. Urinalysis should be obtained in all patients with DAH to evaluate for the presence of a “pulmonary-renal syndrome” which is defined as the presence of DAH plus glomerulonephritis. Glomerulonephritis in turn is characterized by the presence of (i) proteinuria, (ii) microscopic (or gross) hematuria, ideally with dysmorphic, crenulated red blood cells, and (iii) red blood cell casts. Renal insufficiency and renal failure will commonly ensue such that the presence of a pulmonary renal syndrome calls for rapid treatment to prevent permanent renal failure.
Chest radiography is extremely informative and is characterized by diffuse alveolar infiltrates, but is difficult to distinguish from other diseases characterized by an alveolar filling pattern (i.e acute respiratory distress syndrome, congestive heart failure or pneumonia.) The alveolar opacities themselves may vary from a patchy focal process to confluent, diffuse alveolar filling. Still, those cases that initially present with unilateral or lobar infiltrates will usually rapidly progress to diffuse alveolar filling if unrecognized or untreated. HRCT of the chest can confirm the presence of air space filling. While DAH will typically be characterized by patchy, bilateral ground glass opacities ± consolidation with a central and lower lobe predominance, the higher resolution images tend to add only a marginal amount of information over a standard chest radiograph. Finally, although not commonly recognized, repeated bouts of DAH may lead to findings of fibrosis or even obstructive lung disease on chest radiography [8, 9].
Pulmonary function testing may be performed in patients in whom the disease onset is less acute, and in these cases, the diffusing capacity for carbon monoxide (DLCO) may be elevated, or if measured sequentially, may be noted to increase. This increase in DLCO is secondary to the presence of carbon monoxide-avid hemoglobin in the airspaces. However, in patients who present with more acute disease, pulmonary function testing is rarely feasible. Longitudinally, pulmonary function testing can be useful in cases of DAH in which the bleeding may be chronic or recur frequently, such as idiopathic pulmonary hemosiderosis or anti-phospholipid antibody syndrome as these patients may go on to develop obstructive and/or restrictive physiology.
Diagnosis (Table 10.1)
While the presence of DAH may be strongly suggested in a patient with marked hemoptysis, bilateral alveolar infiltrates, anemia and respiratory distress, this classic presentation appears to represent a minority of cases. Ultimately, DAH remains on the differential diagnosis of any patient with bilateral or diffuse alveolar infiltrates, hypoxemia and dyspnea, and in point of fact, autopsy studies have shown that 2–4 % of patients with clinical acute respiratory distress syndrome (ARDS) who died of their disease will be found to have unsuspected DAH [10, 11]. Thus, although pneumonia, heart failure and ARDS are all much more common than DAH, in those cases where the diagnosis is less than certain, bronchoscopy with BAL should be considered.
When performing the procedure, the BAL should always be performed prior to any concomitant procedure such as biopsy to avoid precipitating any confounding bleeding or even bronchoscope trauma. When choosing the anatomic location for the BAL, the operator should choose the areas most involved by chest radiograph, or in diffuse disease, may choose the right middle lobe or lingula so as to optimize the return volumes. The bronchoscope should be advanced until “wedged” or impacted in a segmental or subsegmental bronchus. Once a position has been secured, four to five standard saline aliquots of between 30 and 60 ml should be serially instilled and removed via the bronchoscope up to a total lavage volume of no less than 100 ml and no more than 300 ml (and ideally >30 % of the total instilled volume should be obtained on return to assure the accuracy of differential cell counts) [12]. In DAH, the recovered fluid will become increasingly hemorrhagic from aliquot to aliquot, or at a minimum, will not clear with serial lavage. This bronchoscopic finding is diagnostic of DAH (Box 10.1). Nevertheless, the finding of DAH is not a final diagnosis in and of itself as the general presence of DAH carries an extended differential diagnosis and cannot by itself define the underlying etiology for the DAH.
Box 10.1
Diagnosis of Diffuse Alveolar Hemorrhage
Entities
Diffuse alveolar hemorrhage is diagnosed at the time of bronchoscopy. With the bronchoscope in “wedge position” in a segmental or subsegmental bronchus, four to five standard saline aliquots of between 30 and 60 ml are serially instilled and removed for a total lavage volume of no less than 100 ml and no more than 300 ml. A diagnosis of diffuse alveolar hemorrhage is made when the recovered fluid is identified to be increasingly hemorrhagic from aliquot to aliquot, or at a minimum, does not clear with serial lavage. Alternatively, a diagnosis of DAH may also be made at time of surgical lung biopsy when a pathologic finding of diffuse alveolar hemorrhage is made (red blood cells filling the alveolar spaces.) If a diagnosis of DAH is made at the time of surgical lung biopsy, a concurrent pathologic diagnosis of capillaritis or bland hemorrhage should also be identified
As mentioned above, serologic testing is central to the evaluation of DAH, and in specific cases, serologic studies can confirm a diagnosis without the need for surgical biopsy. In Goodpasture’s syndrome, diagnosis may be confirmed by the presence of serum anti-basement membrane antibodies (ABMAs.) [13] Similarly, serum anti-cardiolipin antibodies (and Russell Viper Venom Time) should be measured to assess for primary anti-phospholipid antibody syndrome [1]. The presence of serum anti-neutrophil cytoplasmic antibodies (ANCA) and/or a positive anti-proteinase-3 or anti-myeloperoxidase enzyme-linked immuosorbant assay (ELISA) will assist with the diagnosis of a primary, small vessel, ANCA-associated vasculitis (AAV) such as granulomatosis with polyangiitis (the entity formerly known as Wegener’s granulomatosis), microscopic polyangiitis, pauci-immune idiopathic pulmonary capillaritis, or Eosinophilic Granulomatosis with Polyangiitis (EGPA), the entity formerly known as Churg Strauss Syndrome [14]. In cases of DAH complicating SLE, the diagnosis of SLE is usually established [3]. However, in cases where DAH is the presenting manifestation, serum testing for low serum complement (specifically C3 and C4), serum antinuclear antibodies, and the presence of anti-double-stranded deoxyribonucleic acid antibodies will help point to the diagnosis. Anti-SS-A (Ro) and SS-B (La) antibodies are less specific, but may be associated with SLE as well as primary Sjogren’s syndrome and scleroderma. Anti-streptolysin O testing is helpful in the identification of post-streptococcal disease, and cryoglobulins and hepatitis serologies are helpful in the assessment of cryoglobulinemia.
Additional testing that is less specific but may be helpful in diagnosis includes a complete blood count, liver function testing, renal function testing, inflammatory markers, urinalysis with sediment examination, and coagulation studies. Furthermore, echocardiography is often required to evaluate for mitral stenosis, severe diastolic dysfunction and other causes of elevated left-sided filling pressures that potentially may cause bland hemorrhage. Additional imaging studies, beyond chest radiography and HRCT, that may yield diagnostic information depending upon the clinical scenario include CT of the sinuses (i.e. to assess for evidence of granulomatosis with polyangiitis), CT/MRI of the brain, and CT of the abdomen and pelvis.
In some cases, surgical lung biopsy may be required to establish the underlying cause if serologic testing or history are unrevealing. The decision to proceed to surgical lung biopsy should not be taken lightly as the procedure, whether done as a less invasive video assisted thoracoscopic procedure or a more invasive thoracotomy, requires general anesthesia and is an invasive surgical thoracic procedure in a moderately or severely ill patient. On the other hand, surgical lung biopsy may be safely accomplished in the hands of an experienced surgeon in the vast majority of cases. Ultimately, the decision to proceed or not to proceed to surgical lung biopsy must take into account a careful weighing of the risks, benefits and alternatives. It should be clear that the biopsy is revealing critical diagnostic information that cannot be obtained in other ways and that this information will affect treatment decisions.
Three broad categories of pulmonary histopathology are associated with DAH, namely (i) capillaritis, (ii) bland hemorrhage and (iii) diffuse alveolar damage with hemorrhage, and the identification of the underlying histopathologic pattern can often be used to focus the differential diagnosis.
Pulmonary Capillaritis
Histology (Fig. 10.3)
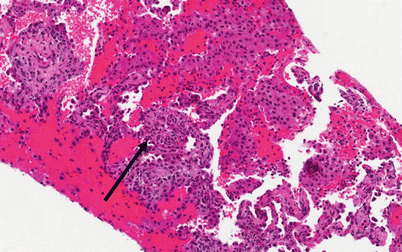
Fig. 10.3
Photomicrograph (20× magnification) of an H&E stained section showing lung parenchyma with diffuse airspace filling by hemorrhage and scattered macrophages. The arrow points to a region of capillaritis in which the alveolar septa are expanded by necrotic neutrophils and karyorrhexitic nuclear debris (Courtesy of Dr. Steven Groshong, Division of Pathology, Department of Medicine, National Jewish Health, Denver, Colorado, USA)
Pulmonary capillaritis, also known as alveolar capillaritis, necrotizing alveolar capillaritis, and neutrophilic capillaritis, is one of the three core histologic pattern that may be associated with DAH. Pulmonary capillaritis is characterized by neutrophils infiltrating the alveolar septa along the pulmonary capillaries with associated nuclear debris and hemorrhage. Capillaritis may, in some cases, also involve other small vessels such as the venules and arterioles. There is associated disruption of the alveolar-capillary basement membrane, and red blood cells, edema fluid, fragmented neutrophils, debris and fibrin leak into the alveolar spaces [15]. The alveolar interstitium itself is broadened by the presence of edema, fibrinoid necrosis, inflammatory cells, and red blood cells. The neutrophils in the interstitium and vessel walls degranulate and undergo apoptosis, and as such, often appear pyknotic and undergo karyorrhexis leaving behind characteristic basophilic nuclear debris. Other features that may occur or be identified on this background pattern of lung injury include: small-vessel thrombosis, organizing pneumonia, and type II alveolar epithelial cell hyperplasia. Lastly, it should be noted that pulmonary capillaritis is a subset of pulmonary vasculitis in which the microcirculation of the lung (alveolar capillaries, arterioles, and venules) is predominantly affected and the larger pulmonary vessels are spared [16].
Etiologies
ANCA-Associated Small Vessel Vasculitis: Microscopic Polyangiitis
Microscopic polyangiitis (MPA) is another of the small vessel ANCA associated vasculitides and has a predilection for the microvasculature of the kidney and the lung. MPA is universally associated with a focal segmental necrotizing glomerulonephritis and is characterized by marked constitutional symptoms. MPA can be differentiated from classic polyarteritis nodosa by the lack of involvement of medium sized blood vessels and the absence of systemic hypertension. Distinguishing MPA from GPA can be difficult, but MPA lacks the granulomatous inflammation seen in GPA, only affects the upper airway in <15 % of patients, and is commonly associated with a peri-nuclear ANCA staining pattern (p-ANCA) rather than a c-ANCA pattern. DAH due to pulmonary capillaritis occurs in up to one-third of patients with MPA and represents by far the most common pulmonary manifestation of the disease. In patients with recurrent bouts of DAH related to MPA, both obstructive lung disease and pulmonary fibrosis have been reported [20, 26].
As mentioned above, a diagnosis of MPA essentially requires that the patient have a focal segmental necrotizing glomerulonephritis. Other common clinical manifestations include arthralgias/arthritis, myalgias/myositis, gastrointestinal disease, peripheral nervous system involvement and cardiac disease. As seen in other AAV, non-specific inflammatory markers are elevated (erythrocyte sedimentation rate and C-reactive protein), and non-specific increases in both serum antinuclear antibodies and rheumatoid factor may be present. Circulating immune complexes may be found in a significant minority of patients with MPA, but tissue localization of circulating immune complexes is rarely seen. ANCA are frequently present in patients with MPA, but less so than with GPA, on the order of 50–75 %. Additionally, 85 % of the ANCAs will have a p-ANCA staining pattern that in turn is more commonly associated with anti-myeloperoxidase (MPO) antibodies [17, 26, 27].
As with GPA, DAH in MPA represents life-threatening disease and is associated with an increased mortality. Indeed, an episode of DAH secondary to MPA is associated with a 30 % mortality. For those patients who do survive, the 1-year and 5-year survival is reduced to 82 and 68 %, respectively [26, 28].
Treatment of DAH secondary to MPA is very similar to GPA and consists of supportive care elements plus glucocorticoids and plasmaphresis followed by cyclophosphamide or rituximab. Additionally, recombinant factor VIIa has been tried at the case report level as a modality by which to control refractory alveolar hemorrhage and unremitting respiratory failure in severe cases of DAH due to MPA [29].
ANCA-Associated Small Vessel Vasculitis: Granulomatosis with Polyangiitis (GPA)
Granulomatosis with polyangiitis is the entity formerly known as Wegener granulomatosis and represents one of the more common etiologies associated with pulmonary capillaritis as well as pulmonary renal syndrome. GPA is one of the AAVand is characterized by granulomatous inflammation of the upper and lower respiratory tract and a necrotizing small vessel vasculitis. While the American College of Rheumatology and Chapel Hill Consensus Conference have developed criteria for the classification of the AAV, these criteria perform poorly when used to diagnose an individual patient. The diagnosis of GPA and the other AAV rests upon the clinician integrating clinical, laboratory, radiographic and pathologic data and making an informed clinical judgment that the data do or do not support a diagnosis of GPA.
DAH is estimated to occur in 5–15 % of patients with GPA. Indeed, the presence of DAH alone should raise the possibility of GPA and the other AAV within the differential diagnosis [17]. DAH can occur as an initial manifestation of the disease or it may occur during an exacerbation of a previously established case. DAH may occur as an isolated finding or in conjunction with other pulmonary manifestations of GPA. In a patient series published by Cordier and colleagues, pulmonary capillaritis was identified in 31 % of open lung biopsies obtained in patients with GPA [18]. The presence of DAH, by definition, represents severe, life-threatening disease and correlates with a considerably increased mortality [19].
As mentioned above, GPA commonly presents with upper airway involvement (>80 %) and may manifest with epistaxis, nasal discharge or crusting, septal perforation, otitis, hearing loss, or subglottic or tracheal stenosis. Similarly, the lower respiratory tract is also frequently involved (>80 %) and patients may manifest with cough, dyspnea, chest discomfort or hemoptysis. Radiographically, patients may have infiltrates, consolidation, nodules, cavities, and/or effusion(s). Extra-pulmonary manifestations will commonly include renal involvement/glomerulonephritis, constitutional symptoms, myalgias, arthralgias/arthritis, cutaneous involvement, ocular involvement, and cardiac manifestations [20].
Anti-neutrophil cytoplasmic antibodies (ANCA) are a hallmark of GPA and contribute to the pathogenesis of AAV. Three distinct ANCA staining patterns have been identified, namely cytoplasmic, peri-nuclear and atypical, and it is the cytoplasmic or c-ANCA that have been most closely associated with GPA. c-ANCA in turn, have been shown to recognize the proteinase-3 (PR3) antigen in the vast majority of cases. 85–90 % of patients with generalized active GPA will be c-ANCA and/or anti-PR3 positive [17]. While ANCA titers correlate with disease activity, a rise in ANCA titers needs to be considered within the context of the full clinical assessment, as a change in ANCA titers alone lacks sufficient sensitivity and specificity for predicting disease relapse to be used as such [21]. Also, it should be noted that while a positive c-ANCA or PR3 is very helpful in diagnosing AAV, a negative test does not exclude GPA or AAV in an individual patient.
Treatment of DAH begins with basic supportive care elements such as a secure airway, oxygen therapy and ventilatory support. Once the patient has been stabilized and the “A, B, Cs” of airway, breathing and circulation have been addressed, and any potential coagulopathic state or bleeding diasthesis similarly addressed, treatment directed towards the underlying precipitating disease may begin.
Treatment of GPA requires the use of immunosuppressive agents (cytotoxic medications and systemic corticosteroids) that carry the risk of serious adverse side effects. As such, the intensity of the immunosuppresion must carefully be titrated to disease activity, and disease activity must be careful assessed in each patient. DAH clearly represents organ and life threatening disease and as such qualifies as “severe” disease that necessitates the use of more aggressive immunosuppressive regimens to control the disease activity.
The initial regimen of choice for both generalized active and severe life-threatening disease had been oral cyclophosphamide plus oral corticosteroids based upon the original National Institutes of Health studies demonstrating the efficacy of this regimen for the induction of disease remission [22]. In 2007, Jayne and colleagues published the MEPEX trial (Randomized Trial of Plasma Exchange or High-Dosage Methylprednisolone as Adjunctive Therapy for Severe Renal Vasculitis) in which patients with severe renal disease were treated with corticosteroids and oral cyclophosphamide and additionally randomized to plasma exchange or high dose intravenous methylprednisolone [23]. Dialysis-independent survival was greater in the plasma exchange group than the intravenous corticosteroid group such that the addition of plasma exchange to corticosteroids and cyclophosphamide has since been recommended for the management of patients with severe renal disease. Whether this same strategy may be applied to patients with DAH was tested in a 20 patient case series, and indeed, this strategy appears to be effective in diffuse alveolar hemorrhage as well [24].
In 2010, the Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis (RAVE) trial evaluated the anti-CD-20 monoclonal biologic rituximab for the management of generalized active and severe AAV and found that rituximab was non-inferior when compared with cyclophosphamide [25]. No significant differences in total or serious adverse events were noted between the treatment groups. Subgroup analysis further showed that rituximab was equally effective with cyclophosphamide for the management of alveolar hemorrhage. Thus, rituximab may be used as a potential alternative to cyclophosphamide in severely ill patients, including those with DAH.
Isolated Pulmonary Capillaritis
Isolated pulmonary capillaritis or idiopathic pauci-immune pulmonary capillaritis refers to a small vessel vasculitis that is confined to the lungs. Some experts liken this entity to a lung-limited MPA. DAH in isolated pulmonary capillaritis may or may not be p-ANCA positive, and while no differences can be discerned between those patients who are ANCA positive and ANCA negative, this may be due to inadequate longitudinal follow-up. In one case series of 29 patients, isolated pulmonary capillaritis was the most common cause of DAH with biopsy proven pulmonary capillaritis, followed by GPA and MPA [30]. In this study, isolated pulmonary capillaritis accounted for 28 % of the cases, and there were no clinical, serologic, or histologic features of an alternative systemic disorder. Clinically, three quarters of patients presented with respiratory distress and half required mechanical ventilation. Despite this, there was an 88 % in hospital survival and an overall favorable prognosis for the group. Isolated pulmonary capillaritis is treated along the same lines as AAV and responds well to standard therapy with corticosteroids and cytotoxic medications [27, 31].
Systemic Lupus Erythematosus
DAH affects only 4 % of patients with SLE, but along with acute lupus pneumonitis represents one of the most devastating pulmonary complications of SLE with a mortality rate approaching 50 %. Histopathologically, DAH due to SLE is associated with pulmonary capillaritis in the vast majority of cases, but bland pulmonary hemorrhage and DAH secondary to diffuse alveolar damage may also be seen. Co-morbid and/or precipitating infectious complications should excluded as a contributing factor to the DAH [3, 32, 33].
As with SLE itself, there is a strong female preponderance in DAH secondary to SLE, and patient are on average in their third to fourth decade. In the majority of cases of DAH associated with SLE, glomerulonephritis is also present at the time of presentation. As with other cases of DAH, patients present with dyspnea, hemoptysis, hypoxemia and respiratory distress/respiratory failure; however, this clinical presentation is common to both DAH and acute lupus pneumonitis (ALP) and distinguishing between these entities can be exceedingly difficult. In point of fact, 20 % of cases of ALP may present with hemoptysis. Still, the majority of DAH cases occur in patients with a known diagnosis of SLE and will frequently have concomitant glomerulonephritis, whereas 50 % of cases of ALP are an initial presentation of SLE. Ultimately, as with most cases of DAH, diagnosis is made at time of BAL. In those patients who undergo biopsy, ALP is characterized by diffuse alveolar damage complicated by hemorrhage and may also have features of organizing pneumonia, but should not demonstrate frank capillaritis [3, 6].
As mentioned previously, mortality rates associated with DAH in SLE are high and have traditionally ranged from 50–90 %, although more recent data suggests that the use of aggressive immunosuppressive treatment, increased recognition of concomitant infections, and advances in the management of critically-ill patients, survival is far better. Negative prognostic factors include the need for mechanical ventilation, the presence of infection, and the requirement for cyclophosphamide therapy [3, 6, 33].
Antiphospholipid Antibody Syndrome
Antiphospholipid antibody syndrome, along with GPA, MPA, idiopathic pauci-immune capillaritis, and SLE, represents one of the more common etiologies of capillaritis. As with the other entities, DAH may be an initial presentation or later complication of the disease. Symptoms again include cough, dyspnea, fatigue, malaise, fever, hemoptysis, hypoxemia, and acute respiratory failure. Thrombocytopenia may be present at the time of the DAH episode helping to focus the differential diagnosis on APLAS along with SLE, disseminated intravascular coagulation, and thrombotic thrombocytopenic purpura. On histology, there is evidence of pulmonary capillaritis with or without concomitant microvascular thrombosis [7, 34].
The management of APLAS is commonly complicated thromboemoblic disease and the need for anti-coagulation. When an episode of DAH occurs in a patient with an established diagnosis of APLAS, the presence of capillaritis and diffuse hemorrhage is further complicated by the presence of therapeutic anti-coagulation (as well as the possibility of concomitant pulmonary thromboemboli.) Nevertheless, it must be recognized that more often than not, it is the capillaritis driving the DAH and controlling the vasculitis is key to achieving therapeutic success. At the same time, diagnosing, treating and/or preventing the thromboembolic manifestations of the disease, as well as controlling the DAH, cannot be ignored. Thus, even in centers experienced in the management of complex autoimmune diseases, the management of these patients is extremely challenging and referral to a center of expertise is recommended when feasible. Nevertheless, first line therapy for DAH associated with antiphospholipid antibody syndrome is intravenous corticosteroids combined with optimal supportive care. Rapid resolution of most cases of DAH is typically seen after treatment with corticosteroids. In cases of catastrophic antiphospholipid syndrome, IVIG or plasma exchange may be added to the intravenous corticosteroids and supportive care. Most recently, case reports describing the use of rituximab for refractory APLAS, including APLAS complicated by refractory DAH, have suggested that this agent may ultimately proven to have a role when more conventional therapies are unsuccessful [1, 34–36].
As with other cases of chronic and/or recurrent DAH, fibrosis and/or obstructive disease may evolve over time. Lastly, it should be noted that in catastrophic cases of APLAS (Asherson’s syndrome), ARDS and multi-system organ dysfunction may develop. In these cases, the pathology will demonstrate diffuse alveolar damage, capillaritis and diffuse small vessel occlusion and obliteration [14, 35].
Goodpasture’s Syndrome (Anti-basement Membrane Antibody Disease)
Goodpasture’s Syndrome, or anti-basement membrane antibody disease, is an autoimmune disorder mediated by antibodies directed against the non-collagenous domain (NC1) of the alpha-3 chain of type IV collagen (and to a lesser degree the alpha-5 chain) found in basement membranes [37]. The majority of patients, approximately 60–80 %, present with a pulmonary-renal syndrome of diffuse alveolar hemorrhage and glomerulonephritis. Indeed, the presence of a true pulmonary renal syndrome helps focus the differential diagnosis upon ABMA disease, GPA, MPA, and SLE. Still, 15–30 % of cases may present with isolated glomerulonephritis, and conversely up to 10 % of patients may present with DAH alone without renal involvement. Interestingly, although type IV collagen is found elsewhere in the body, including skin, eye, and gastrointestinal tract, end organ damage in ABMA disease is limited to the kidneys and lung. DAH represents a major cause of mortality in patients with ABMA disease, and among the competing causes of DAH, ABMA disease has a relatively poorer prognosis. On the other hand, in those cases of ABMA disease in which the pulmonary manifestations are dominant, renal outcomes are better when compared to patients who present with renal disease alone [13, 37].
The clinical presentation of DAH due to ABMA disease is very similar to other cases of DAH of differing etiologies of DAH with the caveat that glomerulonephritis is present in most albeit not all cases. Again, patients will complain of dyspnea, cough, hemoptysis, hypoxemia, and/or constitutional symptoms (fatigue, fever, anorexia, weight loss, arthralgias and myalgias). ABMA disease preferentially affects men more than women (approximately 2:1) and has a predilection for young adults (the average patient age reported ranges between 20 and 30). In those patients with ABMA who develop DAH, a history of smoking is extremely common (50–90 %), although recent viral infection or other inhalation exposures (e.g. hydrocarbons, marijuana, fire smoke, cocaine) may also be seen immediately antecedent to the onset of DAH. In point of fact, it is hypothesized that cigarette smoking (or alternatively infection or other inhalational exposure) plays a pathophysiologic role in the development of DAH either through a secondary injury to the alveolar-capillary unit, facilitating antigen presentation, or allowing ABMAs entry into the lung [13]. On laboratory testing, anemia will commonly be present, and frequently will be accompanied by an elevated blood urea nitrogen or serum creatinine consistent with renal insufficiency. Urinalysis frequently reveals microscopic hematuria, proteinuria, and red blood cell casts diagnostic of glomerulonephritis. Interestingly, 3–7 % of patients will be p-ANCA positive, suggesting the possibility of an overlap syndrome with MPA. ABMA disease has also been shown to have an association with specific human leukocyte antigen (HLA)-DR alleles, specifically HLA-DRB1*1501 [37]. Chest imaging studies, as with other cases of DAH, will show patchy diffuse alveolar infiltrates that appears as ground glass infiltrates or consolidation on HRCT. While it is rare to obtain pulmonary function testing in acutely-ill patients, in more chronic cases or more slowly evolving cases, an increased diffusing capacity of carbon monoxide may be identified.
Diagnosis may be made by identifying the presence of serum anti-basement membrane antibodies (ABMAs) in a patient with a compatible clinical presentation and circulating antibodies may be identified in two thirds to three quarters of patients at time of diagnosis [37]. At the bedside, it may be necessary to make a tentative clinical diagnosis and initiate therapy while awaiting the results of the serologic testing which frequently must be sent to a referral laboratory. Of note, while antibody titers appear to correlate with the severity of the renal disease, no such correlation has been identified with the pulmonary manifestations of the disease. Alternatively, patients may be diagnosed via lung or kidney biopsy and immunofluorescence studies. On light microscopy, the histopathology of the lung in ABMA disease may demonstrate either bland hemorrhage or capillaritis (although the appearance of the capillaritis in ABMA disease tends to lack some of the more aggressive and destructive features seen in MPA or GPA). Similarly, the renal biopsy will demonstrate a focal, segmental, rapidly progressive glomerulonephritis with crescent formation that is indistinguishable from other etiologies of rapidly progressive glomerulonephritis. However, the frozen sections should demonstrate a positive immunofluorescence pattern with a linear, continuous, “ribbon-like” appearance, reflecting antibody that has bound to the basement membrane. This immunofluorescence pattern is distinct from the punctate, patchy staining pattern seen in SLE and the negative or “pauci-immune” pattern associated with the AAV, and hence, is diagnostic for ABMA disease [38].
DAH associated with Goodpasture’s is managed very similarly to DAH (or severe renal failure) secondary to GPA or MPA. Plasmapharesis combined with corticosteroids and a cytotoxic agent (i.e. cyclophosphamide) has been shown to be effective in both of these clinical contexts and is associated with improved mortality and renal recovery [39]. Early diagnosis and prompt institution of therapy is crucial to optimizing outcomes, and as such, it is sometimes necessary to initiate therapy pending a conclusive diagnosis. Ultimately, the similarities in therapeutic recommendations for pulmonary renal syndrome, whether it is due to ANCA associated vasculitis, SLE or Goodpasture’s syndrome combined with the adverse effects associated with delays in therapy makes this approach judicious. Steroids alone, or steroids combined with cytotoxic agents without the use of plasmaphresis do not achieve equivalent results. While there is no definitive data informing the optimal duration of plasmaphresis, the duration of therapy for Goodpasture’s tends to be longer than in AAV, on the order of 10–14 exchanges, or until ABMAs become undetectable. With regard to choice of cytotoxic agent, cyclophosphamide is the most common agent utilized for life threatening alveolar hemorrhage. In more mild cases or as the patient’s conditions improves, azathioprine (and more recently mycophenolate mofetil) have been used [20]. Lastly, rituximab has been proposed by some experts as a potential therapeutic agent for Goodpasture’s syndrome based upon its mechanism of action and the pathogenetic role of ABMAs in the disease; however, beyond a handful of anecdotal reports, there is currently no evidence to support its use and its role in managing ABMA disease awaits further study.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


