Adverse Cardiovascular Drug Interactions and Complications: Introduction
Cardiovascular-targeted pharmacotherapy continues to rapidly evolve as newer agents are sought to fill in gaps in care or to lower adverse reactions of existing therapies. With newer agents, benefits are accompanied with new potential reactions, which can have adverse consequences on patients. This chapter covers important aspects of adverse drug reactions and reviews pharmacokinetics and pharmacodynamics of commonly used drugs with an emphasis on mechanisms. Finally, specific syndromes or cardiac disease states are presented, with focused discussion on newer pharmacologic agents than can present challenges to the clinicians faced with the plethora of choices.
Adverse drug reactions (ADRs) are the fourth leading cause of death in patients hospitalized in the United States.1-3 ADRs are responsible for approximately 1 of every 16 hospital admissions and occur in as many as 20% of hospitalized patients.1,4 The cost of these events in financial terms is staggering; estimates of the financial burden of ADRs in the United States range from $30 billion to more than $130 billion annually.2,3
The World Health Organization defines an ADR as “a response to a drug that is noxious and unintended and occurs at doses normally used in man for the prophylaxis, diagnosis and therapy of disease, or for modification of physiological function.”5 ADRs are commonly classified as either type A (augmented) or type B (bizarre) reactions.6 Type A reactions are predictable based on the pharmacologic characteristics of the agent(s) (ie, drug). In contrast, type B reactions are idiosyncratic and unpredictable. This simple classification system remains in use throughout the literature.7 Most of the ADRs discussed in this chapter are type A reactions because they are fairly common, predictable, and often preventable. In this regard, it is absolutely critical that clinical providers including both prescribers and dispensers of drugs remain vigilant of the potential for dangerous, even lethal, ADRs in their treated patient populations, particularly if the patients have cardiovascular disease.
Patients with heart disease represent a population who are at particularly high risk for ADRs. Certain cardiovascular disease states such as heart failure can influence drug metabolism and elimination by altering end-organ perfusion.8 Patients with heart disease are often elderly. Advanced age is associated with higher ADR risk due to age-related alterations in renal and hepatic function, the presence of multiple medical comorbidities, and a high prevalence of polypharmacy.9 Dementia may influence medication compliance, and confusion regarding the indications and doses of prescription drugs is common among older patients, particularly when they receive care from multiple providers.10-12 ADR risk has been shown to increase exponentially with the number of medications prescribed and correlates closely with the total number of drugs taken by an individual patient.13 For example, a patient taking seven medications has the potential for 6 + 5 + 4 + 3 + 2 + 1 = 21 possible drug-drug interactions. The average nursing home patient takes seven medications, and most of these are used to treat cardiovascular diseases.14 Lastly, patients with heart disease often require periodic hospitalization, and medication changes at the time of hospital admission or discharge may contribute to ADR risk.13 Patients who have been hospitalized are at highest risk after discharge when faced with old medications at home in addition to newly prescribed ones, contributing to the confusion on which to take. This is one reason why the Joint Commission has made drug reconciliation at discharge a critical performance measure for quality. Communication of the hospital teams with the primary care provider in charge of the patient can do much to resolve this long-standing dilemma associated with our fragmented health care system.
This chapter provides an overview of the adverse reactions and interactions associated with the use of cardiovascular drugs. It is organized by disease state rather than by drug class to highlight the clinical relevance of each interaction. Newly approved drugs have been emphasized, with more information provided than those for well-established ones. It is worth noting that the ADR potential for an individual drug can vary depending on the indication for its use and the patient using it. For example, when flecainide is used to treat a supraventricular tachycardia in an otherwise healthy 40-year-old patient, the drug is generally efficacious and well tolerated; however, the same dose of flecainide used to treat nonsustained ventricular tachycardia in a 75-year-old patient with an ischemic cardiomyopathy is potentially lethal and therefore contraindicated.
Clinical Pharmacology
Drug interactions can be classified as being either pharmacokinetic or pharmacodynamic. Pharmacokinetic interactions alter the delivery of a drug to its site of action. Pharmacodynamic interactions alter the effect of a drug at its site of action. Clinically relevant interactions between drugs can be pharmacokinetic, pharmacodynamic, or both. For example, patients who are taking both amiodarone and digoxin are at increased risk for symptomatic bradycardia. Amiodarone inhibits the clearance of digoxin.15 This increases the bioavailability of digoxin and results in greater digoxin delivery to cardiac tissue, a pharmacokinetic interaction. Amiodarone also blocks the AV node. This augments the effect of digoxin on AV nodal conduction, a pharmacodynamic interaction.
The effective delivery of a drug to its biological target depends on its absorption, distribution, metabolism, and elimination. Pharmaco-kinetic interactions may occur at any of these steps, leading to either amplification or diminution of the drug’s primary effect or its side effects.
Absorption determines drug bioavailability, defined as the degree to which a drug becomes available at its site of biologic action. Most orally administered drugs are absorbed by the small intestine, so agents that influence gastrointestinal metabolism, motility, or pH have the potential to interact with numerous drugs. Drugs that increase GI motility (metoclopramide) tend to reduce the bioavailability of other drugs, whereas those that decrease motility (anticholinergic drugs) may increase drug bioavailability by allowing for a longer period of absorption.16 Drugs may also bind to one another in the GI tract and reduce bioavailability; for example, this can occur when digoxin is concomitantly administered with antacids.17 The bioavailability of other agents may also be altered by food ingestion. It is important for the clinician to understand the effects of food ingestion (eg, timing of meals or composition of meals as with a fatty meal) to appropriately advise patients.
Once absorbed (or injected), many drugs bind to high-affinity sites on plasma proteins such as albumin and establish some degree of equilibrium between free and protein-bound states. The volume of distribution (Vd) is a theoretical measure that reflects how well a drug is removed from the plasma and distributed in tissue and is related to the serum concentration of a drug by the formula Vd = D/C, where D is the drug dose and C is the serum concentration. The pharmacologic effect of a drug is proportional to the concentration of the drug in the free state. The extent to which a drug binds plasma proteins and equilibrates between the free and bound states varies depending on the biochemical characteristics of the drug. Alterations in protein binding can influence the delivery of a drug to its site of action by influencing the proportion of free drug in the plasma. However, the clinical relevance of changes in drug distribution is frequently offset by reciprocal changes in drug elimination. For example, when digoxin and heparin are administered simultaneously, heparin has been shown to displace digoxin from protein-binding sites, thereby increasing the concentration of free digoxin.18 Although this may transiently increase the delivery of digoxin to cardiac tissue, the increase in free digoxin may be accompanied by a concomitant increase in digoxin elimination by the kidneys.
Drug distribution may also be influenced by the behavior of membrane transport proteins located in cells that comprise the blood-tissue interface of various organs. P-glycoprotein (P-gp) is an ATP-dependent efflux membrane transporter that was originally isolated from multidrug-resistant cancer cells.19 P-gp has also been isolated from healthy human tissue including the small intestine, liver, and blood-brain barrier, where it is thought to regulate the passage of xenobiotic substances in and out of cells. Several drugs appear to depend on P-gp for intracellular transport, most notably digoxin.20 Cardiac drugs known to interact with digoxin, such as verapamil and amiodarone, have also been shown to inhibit the activity of P-gp.21 Thus membrane transport protein modulation represents another avenue toward pharmacokinetic drug-drug interactions in humans.
Most drugs undergo at least some degree of hepatic metabolism. The liver receives absorbed drugs from the small intestine via the portal vein and through a series of enzymatic reactions converts these relatively hydrophobic agents into water-soluble compounds that are more readily eliminated from the body. Hepatic metabolism consists of two phases, biotransformation and conjugation (Fig. 95–1). During biotransformation (phase I), drugs are rendered more hydrophilic by oxidation, reduction, or hydrolysis. Phase I is typically followed by conjugation (phase II), during which drugs receive a molecular attachment such as a glucuronate that can facilitate drug transport within the body. Most drug-drug or drug-nutrient interactions involve the induction or inhibition of phase I metabolic enzymes. The majority of these interactions involve cytochrome P450 (CYP) isozymes.
Figure 95–1.
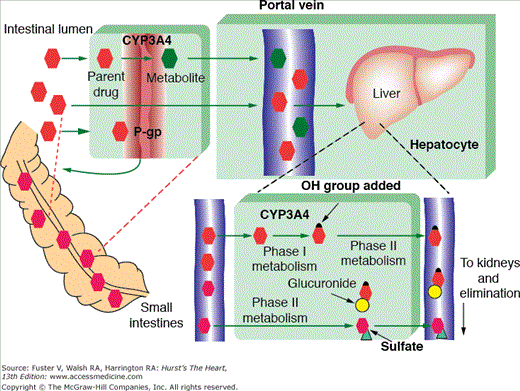
Drug metabolism. Parent drugs enter the portal circulation using protein transport system such as P-glycoprotein (P-gp). Phase 1 metabolism facilitates systemic drug distribution and involves hydrolysis, reduction, or oxidation by enzymes such as cytochrome P450 (CYP) 3A4. Phase 2 metabolism facilitates drug elimination and involves sulfonation and glucuronidation. Reproduced with permission from Page et al.83
CYP is an iron-dependent oxidative enzyme found within the sarcoplasmic reticulum of hepatocytes and, to a lesser extent, the small intestine, kidneys and brain. Although more than 30 CYP isozymes have been identified, 6 of them are responsible for more than 90% of human oxidative drug metabolism, and 1, CYP3A4, is involved in the oxidation of half of all drugs.22 CYP inhibition or induction causes the serum concentrations of substrate drugs to increase or decrease, respectively. In addition, many drugs are metabolized by more than one CYP isozyme. CYP induction also increases with hepatic blood flow and decreases with age.22,23Table 95–1 lists common interactions with the P450 system.24
| CYP Isozyme | ||||||
|---|---|---|---|---|---|---|
| Function | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP2E1 | CYP3A4 |
| Substrate | Caffeine Clozapine Cyclobenzaprine Fluvoxamine Imipramine Mexiletine Olanzapine Pimozide Propranolol Tacrine Theophylline Warfarin | Amitriptyline Citalopram Clomipramine Cyclophosphamide Diazepam Imipramine Lansoprazole Nelfinavir Omeprazole Phenytoin | Amitriptyline Celecoxib Diclofenac Flurbiprofen Ibuprofen Losartan Naproxen Phenytoin Piroxicam SMX Tolbutamide Warfarin | Amitriptyline Clomipramine Codeine Desipramine Dextromethorphan Imipramine Metoprolol Nortriptyline Oxycodone Paroxetine Propafenone Risperidone Thioridazine Timolol Tramadol Venlafaxine | Acetaminophen Chlorzoxazone Dapsone Enflurane Ethanol Halothane Isoflurane Isoniazid | Aliskiren Alprazolam Astemizole Buspirone CCB Carbamazepine Cisapride Cyclosporine Doxorubicin Erythromycin Etoposide Fentanyl HIV PI Iphosphamide Lovastatin Midazolam Pimozide Quinidine Quinine Simvastatin Tacrolimus Terfenadine Triazolam |
| Inhibitor | Cimetidine Ciprofloxacin Citalopram Diltiazem Enoxacin Erythromycin Fluvoxamine Mexiletine Ofloxacin Tacrine Ticlopidine | Cimetidine Felbamate Fluoxetine Fluvoxamine Ketoconazole Lansoprazole Omeprazole Paroxetine Ticlopidine | Amiodarone Fluconazole Fluoxetine Fluvastatin Isoniazid Metronidazole Paroxetine Phenylbutazone SMX/TMP Sulfaphenazole Ticlopidine | Amiodarone Chlorpheniramine Fluoxetine Haloperidol Indinavir Paroxetine Propafenone Quinidine Ritonavir Sertraline Thioridazine Ticlopidine | Disulfiram Water cress | Amiodarone Cimetidine Conivaptar Cyclosporine Danazol Diltiazem Fluconazole Grapefruit juice HIV PI Itraconazole Ketoconazole Macrolides Miconazole Nefazodone Omeprazole Quinidine Ritonavir Verapamil |
| Inducer | Carbamazepine Tobacco | Carbamazepine Norethindrone | Phenobarbital Rifampin Secobarbital | Ethanol Isoniazid | Tobacco | Carbamazepine Rifabutin Rifampin Ritonavir |
Most drugs are eliminated by the kidneys, either through glomerular filtration, active tubular secretion, or passive tubular reabsorption.18 Substances that interfere with the function of the kidneys at any of these levels may precipitate a pharmacokinetic drug interaction. P-glycoprotein, mentioned previously, is found in secretory organs such as the liver, kidneys, and small intestine. Inhibition or induction of these proteins may also influence drug elimination.
Pharmacodynamic interactions occur commonly in the treatment of cardiovascular disease because many cardiac drugs have overlapping physiologic effects. Heart failure therapy provides a useful example. Contemporary management of New York Heart Association (NYHA) class III heart failure recommends treatment with a β-blocker, angiotensin-converting enzyme inhibitor/angiotensin II receptor blocker, and aldosterone receptor antagonist in some cases, in others a combination of hydralazine and nitrates, and in many cases a loop diuretic. All of these drugs can reduce blood pressure, so the development of symptomatic hypotension in such a patient would be considered a pharmacodynamic interaction. Although the clinician should endeavor to avoid antagonistic interactions such as drug-induced hypotension, some pharmacodynamic interactions are therapeutically synergistic. For example, the diuretic metolazone enhances sodium delivery to the loop of Henle and increases the diuretic effectiveness of loop-acting drugs, such as furosemide.25 Pharmacodynamic events may also involve additive medication side effects, though these need not be detrimental.
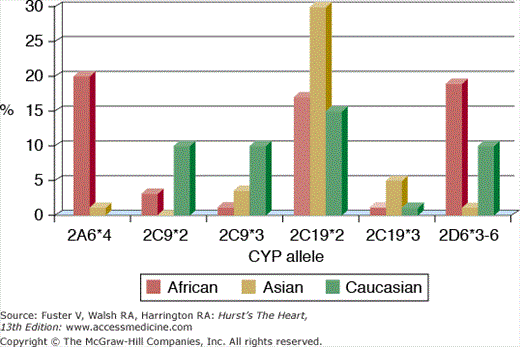
Human genetic diversity influences the pharmacokinetics and pharmacodynamics of cardiovascular drugs. The frequency of genetic polymorphisms involving CYP 450 isozymes varies by ethnic group (Fig. 95–2), although the clinical relevance of these polymorphisms is not uniform. For example, one-third of Caucasians carry at least 1 variant allele for the gene encoding CYP2C9, which is involved in the metabolism of warfarin.26 The presence of this polymorphism increases the anticoagulant effect of warfarin, and affected individuals require lower doses and more frequent monitoring. However, although patients with deactivating polymorphisms involving CYP2D6 experience up to a five-fold increase in serum metoprolol levels compared with unaffected individuals, adverse events and poor tolerability generally do not occur.5 Polymorphisms involving the expression of α- and β-adrenergic receptor subunits appear to influence responsiveness to antihypertensive drug therapy.2,27 Susceptibility to drug-induced torsade de pointes may be affected by polymorphisms involving ion channel genes.28 Understanding how to incorporate knowledge of an individual’s genotype into prescribing pharmacotherapy remains a work in progress that requires randomized clinical trials to best delineate the advantages of such a personalized medicine approach.
Dietary behavior may influence the pharmacokinetics and pharmacodynamics of certain cardiac drugs. Grapefruit juice is a popular beverage and potent CYP3A4 inhibitor that significantly increases serum levels of commonly used drugs such as simvastatin and felodipine.29,30 The anticoagulant effects of warfarin may be substantially reduced in patients who consume vitamin K–rich foods such as lettuce, spinach, avocado, asparagus, and canola oil.31 Herbal remedies have become enormously popular in recent years; one-half of Americans surveyed have used herbal products at least once, and one-quarter report regular use. Relatively few adverse herb-drug interactions have been described in the literature, but this may reflect the fact that patients seldom report the use of herbal products and physicians seldom ask.32Hypericum perforatum (St. John’s wort), a popular herbal remedy for treating depression, decreases plasma levels of digoxin, possible due to P-glycoprotein transport induction.33H. perforatum also reduces cyclosporin levels and has been implicated as a contributor to acute rejection of a transplanted heart.34 Black licorice (Glycyrrhiza glabra) can raise blood pressure and pharmacodynamically competes with aldosterone antagonists for binding at the mineralocorticoid receptor.35
The elderly are particularly vulnerable to drug-drug interactions. Drug pharmacokinetics may change with advancing age for several reasons. Percent body fat tends to increase with age and may increase the volume of distribution of fat-soluble drugs. Conversely, cachexia can increase serum levels of drugs with a large volume of distribution such as digoxin.36 Therefore, in the elderly, digoxin toxicity can occur at lower doses, and levels should be monitored. If an elderly patient is anorectic, certain medications may be more rapidly absorbed in the absence of food ingestion. Although hepatic metabolism is relatively unaffected by age in the absence of overt liver disease, alterations in hepatic blood flow may reduce the first-pass metabolism of highly extracted drugs. Hepatic metabolism may never be more pertinent than in patients with heart failure, especially among patients with right heart failure and peripheral congestion. Glomerular filtration and renal tubular secretion decreases with increasing age and may influence drug clearance. Drug pharmacodynamics are also influenced by age. β-Adrenergic receptor sensitivity decreases with age and may reduce the efficacy of β-blocker therapy.37,38 However, aging often accompanies other comorbidities, and age should not a priori change medication target doses, although up-titration should be done with caution and care. Finally, age-related changes in baroreceptor reflex sensitivity may increase the risk of orthostatic hypotension in elderly patients taking antihypertensive medications, particularly if somewhat volume depleted.36
Cigarette smoking may influence the metabolism of cardiovascular drugs by increasing phase I hepatic enzyme activities. Heavy smoking has been shown to increase the activity of CYP2D6 four-fold when compared with nonsmokers.39 Increased CYP1A2 activity has also been observed in male smokers. Clinicians should be mindful of the effects of smoking cessation on cardiovascular drugs that are metabolized by CYP1A2.40
Heart Failure and Transplant
Heart failure is the most common reason for hospitalization among Medicare recipients, and the number of patients treated for heart failure is expected to increase as the US population ages.41 Clinical trials of new agents for the treatment of heart failure historically use the previously proven drug as background therapy upon which to test the newest agent. Therefore, heart failure is the classic “add-on-therapy” syndrome, putting patients at risk for the adverse complications of multidrug therapy. In addition, the majority of patients with heart failure have comorbid conditions that are likely to require their own pharmacotherapies. Furthermore, the impaired cardiac output that is characteristic of heart failure may slow gastrointestinal transit time, affecting drug absorption and influencing hepatic blood flow. Renal dysfunction is also common in heart failure and may affect drug elimination. Therefore, heart failure patients should be considered at high risk for ADRs. The following sections highlight some of the more commonly encountered ADRs in heart failure.
Heart failure is common among patients with advanced diabetes.42-44 The American College of Cardiology/American Heart Association Guidelines for the treatment of chronic heart failure have identified diabetes as one of the risk factors for stage A heart failure.45 Therefore, the concomitant disorders of heart failure and diabetes are commonly seen by many clinicians. Among the advances of diabetes treatment has been the emergence of the thiazolidinediones (TZDs). These agents improve insulin sensitivity for more effective glycemic control. Troglitazone, the first on the market, was withdrawn due to liver toxicity. Rosiglitazone and pioglitazone are approved as monotherapy and in combination with other oral hypoglycemic agents; pioglitazone is also approved in combination with insulin. Other potential beneficial effects of these agents make them particularly attractive in treatment of cardiovascular disorders. When compared with glyburide, rosiglitazone may decrease diastolic blood pressure.46 Pioglitazone has been shown to suppress in-stent neointimal proliferation, which may be related to improvements in endothelial dysfunction. Particularly in patients with an ischemic cardiomyopathy, pioglitazone may be ultimately beneficial,47,48 though this needs to be established in appropriately designed randomized clinical trials. Despite these potential benefits, the TZDs can cause fluid retention and worsening edema in patients with heart failure. The fluid retention appears to be worse with concomitant insulin use.42,49 The mechanism for this is unclear, but warnings about TZD use in patients with NYHA class III and IV heart failure appear in the prescribing information. Therefore, these agents need to be used with caution in advanced heart failure, and patients should be closely monitored for weight gain. Additional diuretic use is frequently required. In spite of the potential benefits of these agents, they may need to be discontinued in patients for whom increasing volume control becomes a difficult problem.
The publication of the landmark Randomized Aldactone Evaluation Study (RALES) trial, which showed that the addition of an aldosterone antagonist to standard therapy dramatically improved survival in severe heart failure patients, was followed by an unprecedented increase in spironolactone prescription.50-53 However, the extension of spironolactone use to patients with less severe heart failure and outside the careful monitoring environment of a clinical trial has resulted in an alarming increase in the occurrence of hyperkalemia and its consequences.53 The advent of the angiotensin II receptor blockers (ARB) and their use as “add-on” therapy in patients treated with angiotensin-converting enzyme inhibitors (ACEI) significantly increases the likelihood of pharmacodynamic drug interactions with spironolactone, making serious hyperkalemia even more likely.54 The addition of spironolactone in such patients must be done with great caution, and close monitoring of renal function and potassium is mandatory. Until additional evidence-based data are published that suggest otherwise, the use of spironolactone in heart failure should be limited to patients who fit the entry criteria of the RALES trial.
Eplerenone is a selective aldosterone antagonist similar to spironolactone. In contrast to spironolactone, the activity of eplerenone at progesterone, androgen, and glucocorticoid receptors is significantly reduced. Eplerenone is also less potent at mineralocorticoid receptors compared with spironolactone. However, eplerenone is significantly more selective for the mineralocorticoid receptor over the other receptors. This selectivity leads to a lower incidence of gynecomastia, breast pain, and impotence.55
Eplerenone is metabolized by CYP3A4 and caution should be exercised with other inhibitors or inducers of CYP3A4. In the EPHESUS trial, addition of eplerenone to standard therapy in patients post acute myocardial infarction complicated by left ventricular dysfunction (left ventricular ejection fraction ≥40 or presence of diabetes) and heart failure was associated with a reduction in all-cause mortality of 15% and reduction in combined end point of cardiovascular mortality and cardiovascular hospitalization of 13% (Fig. 95–3). Patients with elevated serum creatinine were excluded.56,57 Concerns about renal insufficiency and the risk of hyperkalemia are similar to those with other aldosterone antagonists. The drug is contraindicated in severe renal or hepatic impairment. Eplerenone is approved for the treatment of hypertension alone or with other agents and for improving survival of stable patients with left ventricular systolic dysfunction (left ventricular ejection fraction ≥40%) and heart failure after an acute myocardial infarction.
Figure 95–3.
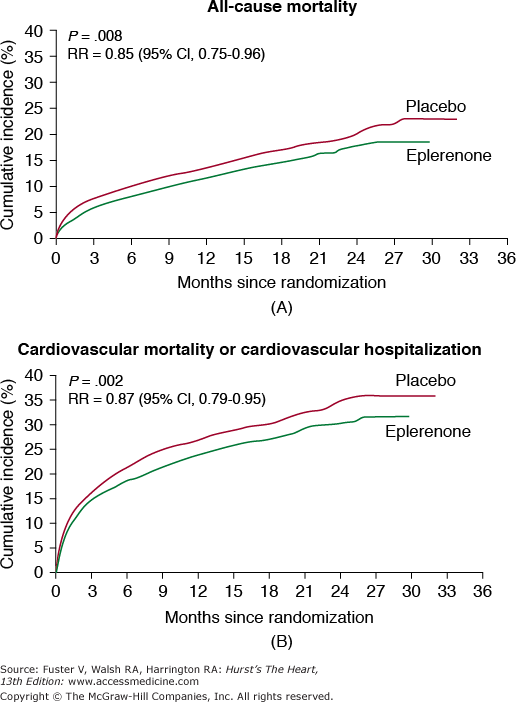
When compared with placebo, eplerenone showed 15% reduction in overall mortality (A) and 13% reduction in cardiovascular death or hospitalization for cardiovascular events (B). Reproduced with permission from Pitt et al.57 Copyright © 2003 Massachusetts Medical Society. All rights reserved.
Once thought to be contraindicated in patients with left ventricular systolic dysfunction, certain β–blockers have since been shown to dramatically reduce morbidity and mortality in patients with heart failure.58-60 The benefits extend from patients with NYHA class II symptoms to those with more advanced disease (class III and IV). In spite of these significant trials, the administration and dosing of β-blockers requires some careful attention to avoid giving “the right drug at the wrong time.” β-Blockers are negative inotropic agents, and their first effect is to reduce ventricular contractility.61 These are the pharmacologic effects of the drug. However, their biologic effects (possible reverse remodeling) are time-dependent. Therefore, heart failure patients should be carefully examined to assure euvolemia before initiating β-blocker therapy. If administered to a volume overloaded patient, β-blockers may contribute to volume excess and precipitate heart failure decompensation. A mild increase in intravascular volume is common when β-blockers are started or when doses are increased. This volume increase is usually transient and can often be controlled with additional diuretics, though patients should be followed at frequent intervals until their volume is adequately controlled. Caution should be used when beginning these agents in patients with advanced heart failure, although their use in this population is strongly recommended based on the evidence from the clinical trials. Appropriate timing and dosing will allow most heart failure patients to initiate and continue these lifesaving drugs. Current American Heart Association/American College of Cardiology Guidelines for the treatment of heart failure recommend initiation of these agents before discharge from the hospital. If doing so, early follow-up is recommended.
These powerful drugs have been proven to increase survival and decrease hospitalizations in patients with NYHA class II to IV heart failure.62,63 In spite of these benefits, clinicians continue to both underuse and underdose these agents. Some of the concerns arise from inappropriate timing of administration. As with β-blockers, accurate volume assessment is critical when administering ACEIs for heart failure. Patients with clinical hypovolemia, frequently due to overly aggressive diuresis, may experience acute renal failure when ACEIs are initiated or increased. However, these changes are usually transient and rather than remove the ACEI, the diuretics should be discontinued or reduced. Avoiding the impulse to rapidly and aggressively diurese will usually allow successful introduction and up-titration of these important agents. A similar rationale should be applied to ARB administration in heart failure, either alone or in combination with ACEI.64
The fixed-dose vasodilator combination of isosorbide dinitrate and hydralazine is indicated for treatment of heart failure in African Americans. The African-American Heart Failure Trial (A-HeFT) showed that addition of this drug to optimized background heart failure therapy in African American patients was associated with a decrease in mortality of 43%, while also decreasing hospitalization and improving quality of life. The study was prematurely stopped due to benefit in the patients on the active drug arm.65 In a post hoc analysis, the benefit from isosorbide/hydralazine was similarly observed in both sexes.66 Use of isosorbide dinitrate/hydralazine is contraindicated with concurrent use of phosphodiesterase-5 inhibitors. The hydralazine component can in rare cases cause a drug-induced lupus-like syndrome. Isosorbide/hydralazine can also cause reflex tachycardia and result in increased oxygen demand on ischemic hearts if not used concomitantly with a β-blocker. Finally, caution should be used in patients with hypertrophic cardiomyopathy because the drug can worsen obstruction by decreasing preload. Whether other forms of nitrates such as the mononitrates can be substituted for isosorbide dinitrate is a matter of controversy because these alternatives have not be subjected to the same rigors of efficacy evaluation in a large clinical trial. Currently, hydralazine dosed separately with a nitrate is frequently administered in lieu of the fixed-dose drug. Each combination tablet contains 20 mg of isosorbide dinitrate and 37.5 mg of hydralazine hydrochloride. The maximum recommended dose is 2 tablets three times a day (ie, six tablets daily).
Concerns have been raised about the loss of effectiveness with the concomitant use of ACEIs and aspirin. This issue is particularly relevant because more than 50% of patients with heart failure have coronary artery disease and are likely to be prescribed aspirin as a primary or secondary therapy agent. Much of the controversy surrounding aspirin has been generated by review of large randomized trials.67-69 This important question was one of the objectives of the Warfarin and Antiplatelet Therapy in Chronic Heart Failure (WATCH) trial.70 The trial was stopped due to poor recruitment and futility. However, a retrospective analysis did show 27% fewer patients hospitalized for worsening heart failure in the warfarin group compared with the aspirin group (P = .01), as well as a 31% lower overall rate of heart failure hospitalizations.71 In spite of this observation, the aspirin group did not have increased adverse outcomes such as subsequent death, myocardial infarction, or stroke. This issue remains unresolved. The Warfarin Versus Aspirin in Patients With Reduced Cardiac Ejection Fraction (WARCEF) trial is an ongoing National Institutes of Health–funded study of aspirin versus warfarin in patients with heart failure.72 WARCEF will evaluate events related to aspirin use in patients with heart failure, and recruitment has ended enrollment, with the results pending. Further discussions concerning ACEI and nonsteroidals are noted below in the Hypertension section of this chapter.
Conivaptan and tolvaptan are vasopressin receptor antagonists, a class of drugs that inhibit vasopressin V2 receptors in the basolateral membrane of the collecting duct cells of the kidneys (tolvaptan) or both the V2 and V1 receptors (conivaptan). V1 receptors found in the myocardium and blood vessels are thought to be responsible for peripheral and coronary vasoconstriction, as well as cell growth and increased intracellular calcium. In the kidneys, these drugs lead to a net aquaretic effect73 (Fig. 95–4).74 The net water loss achieved by this class of medications has been associated with a rise in plasma sodium levels. Conivaptan is approved for the treatment of euvolemic or hypervolemic hyponatremia in hospitalized patients. Conivaptan is available only in intravenous form and is a strong inhibitor of CYP3A4 and therefore can increase the levels of many cardiac medications, including digoxin, eplerenone, and everolimus.
Figure 95–4.
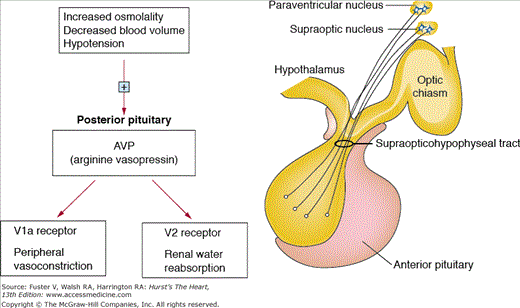
Vasopressin release by the posterior pituitary is stimulated by increased osmolality, hypotension, and hypovolemia. The main two effects of vasopressin include vasoconstriction via V1a receptors and water retention via V2 receptors in the kidneys. Adapted with permission from Oghlakian and Klapholz.74
Recently, the US Food and Drug Administration (FDA) approved tolvaptan, a selective vasopressin receptor antagonist, for the treatment of patients with clinically significant hypervolemic and euvolemic hyponatremia in patients with heart failure, cirrhosis, and the syndrome of inappropriate anti-diuretic hormone (SIADH). Tolvaptan is metabolized in the liver via the CYP3A4 system. Its use is contraindicated in patients on strong inhibitors of CYP3A4. Tolvaptan can also lead to increases in potassium, which should be monitored, especially considering that heart failure patients are usually on concomitant ACEI/ARB and possibly aldosterone antagonists. Tolvaptan is available orally but tested primarily in hospitalized patients (SALT 1 and 2 and EVEREST). In the EVEREST trial, which tested tolvaptan against placebo in a group of heart failure patients admitted with signs and symptoms of volume overload, tolvaptan decreased body weight more than placebo but failed to impact the primary end point of all-cause mortality or cardiovascular mortality or heart failure hospitalization.75
α-Blockers have been used for years to treat hypertension. Prazosin was developed for hypertension, but its presumed effect of afterload reduction made it an attractive drug for the treatment of heart failure. Reports of tachyphylaxis and fluid accumulation made the drug difficult to use in heart failure patients. However, when tested prospectively in the VHeFT trial, the drug was no better than placebo on mortality in heart failure patients.76 More recently, the α-blocker doxazosin was tested as an arm of the ALLHAT trial and stopped early due to onset of heart failure.77 Doxazosin is now being used frequently for symptomatic treatment of prostatic hypertrophy and urinary hesitancy. In patients with advanced heart failure, this drug must be used cautiously because of its inherent fluid accumulation potential. If used together, diuretics should be adjusted as needed.
The role of the renin-angiotensin-aldosterone system blockers has been evaluated in heart failure with preserved ejection fraction (HF-PEF) patients.78 To date, none of the clinical trials showed any significant morbidity or mortality benefits of such agents in this population, including CHARM-preserved (candesartan)79 PEP-CHF (perindopril),80 and I-PRESERVE (irbesartan).81
Cardiac transplant recipients are at unique danger of drug-drug interactions as a result of general unfamiliarity with immunosuppressive drugs by most nontransplant clinicians and the narrow therapeutic window these drugs possess. After transplant, patients require a variety of immunosuppressant and nonimmunosuppressant drugs. Interactions may be inevitable because of the complexities of the medication regimens, and very careful thought is warranted by clinicians caring for this group of patients. Interactions with cyclosporine are numerous and can lead to increased serum levels with subsequent hypertension and renal failure or, conversely, to graft rejection if levels drop significantly. It would be challenging, if not impossible, to list each and every ADR for this population. This section selects the most common and potentially dangerous ADRs involving transplant patients. Although many of the references reviewed in this section pertain to renal transplant primarily, we have extrapolated these findings to the cardiac transplant population. However, it is critical to note that whenever a new drug is introduced to the cardiac transplant patient’s regimen, it is prudent to consider possible drug-drug interactions. Consultation with a specialist expert in the care of the cardiac transplant patient may be indicated, especially if medication changes are numerous and/or complex.
Cyclosporine (CSA) and tacrolimus (TAC) belong to the family of calcineurin inhibitors that undergo metabolism via hepatic and intestinal CYP3A4. Oral CSA and TAC have incomplete, irregular absorption that varies from patient to patient. Table 95–2 depicts a variety of interactions with commonly used agents after transplant. It is important to remember that after transplant, hypertension and hyperlipidemia are common. Therefore, patients will often require ≥1 antihypertensives medications in addition to lipid-lowering therapy. Careful monitoring of CSA and tacrolimus levels are critical to avoid rejection or alternatively excessive levels and side effects.
| Drug Class | Examples | Effect | Onset | Management |
|---|---|---|---|---|
| Antihypertensives | Amlodipine | Increased TAC/CSA effect | Delayed | Monitor TAC/CSA levels 3 times per week. |
| Diltiazem | Reduce TAC/CSA dose by 20%-50% with diltiazem or verapamil. | |||
| Felodipine | ||||
| Nifedipine | ||||
| Verapamil | ||||
| Lipid-lowering agents | Atorvastatin | Increased statin effect with risk for myopathy or rhabdomyolysis | Delayed | Use lowest possible statin dose |
| Fluvastatin | Consider fluvastatin or pravastatin | |||
| Lovastatin | Use lowest possible ezetimibe dose | |||
| Pravastatin | Increased ezetimibe effect | Monitor TAC/CSA 2-3 times weekly for first week then weekly for one month | ||
| Rosuvastatin | Decreased TAC/CSA effect | |||
| Simvastatin | ||||
| Ezetimibe | ||||
| Gemfibrozil | ||||
| Fenofibrate | ||||
| Antiplatelet agents | Clopidogrel Ticlopidine | Decreased clopidogrel metabolite | Delayed | Monitor TAC/CSA levels closely for several months. Monitor for abnormal clotting |
| Azole antifungals | Clotrimazole | Increased TAC/CSA effect | Delayed | Monitor CSA/TAC levels 2-3 times for first week |
| Fluconazole | Increased TAC/CSA effect | Delayed | Monitor CSA/TAC levels 2-3 times for first week | |
| Itraconazole | Increased TAC/CSA effect Nephrotoxicity | Rapid | Monitor CSA/TAC levels 2-3 times for first week; reduce initial dose of CSA/TAC by 50% | |
| Ketoconazole | Increased TAC/CSA effect Nephrotoxicity and hepatotoxicity | Rapid | Monitor CSA/TAC levels 2-3 times for first week; reduce initial dose of CSA/TAC by 50% | |
| Monitor renal and hepatic functions closely |
Diltiazem is a commonly used antihypertensive in this population due to a positive effect on transplant arteriopathy in a small but randomized study.82 Diltiazem inhibits both CYP3A4 and P-gp and raises CSA levels 1.5- to 6-fold, requiring a reduction in CSA dosing by 20% to 75%. A similar reduction is necessary for TAC.83,84 Dihydropyridine calcium channel blockers also inhibit the CYP3A4 system and may potentially interaction with CSA and TAC. In one study, diltiazem, at a dose of 120 mg daily, increased sirolimus (SIR) levels in healthy subjects.85 The increase in SIR bioavailability was attributed to inhibition of CYP3A4 by diltiazem. Thus far, by observation from efficacy data, everolimus levels have not been affected by potential CYP3A4 inhibitors such as the dihydropyridines, diltiazem, or verapamil.86
Atorvastatin, simvastatin, and lovastatin are all substrates for CYP3A4 that may interact pharmacokinetically with CSA and TAC, resulting in myopathy or even rhabdomyolysis.87 Fluvastatin is metabolized primarily by CYP2C9 and pravastatin through other pathways that do not fully involve the CYP enzyme system. Rosuvastatin, now approved, exhibits minimal metabolism via the CYP enzyme system.84 With the exception of fluvastatin, all the statins have been associated with rhabdomyolysis when used concomitantly with CSA.87 When dosing statins, the lowest effective dose should be initiated, and monitoring for myopathy should follow. If rhabdomyolysis occurs, the lipid-lowering agents should be discontinued. Pravastatin appears to have the least accumulation in the presence of CSA.88 When in doubt regarding these interactions, a consultation with a pharmacist may be warranted.
Antibiotics are frequently prescribed for patients after cardiac transplant, particularly during the first posttransplant year, when the delicate balance between rejection and over-immunosuppression exists. Depending on the structure and metabolism of antibiotics, both CSA and TAC are likely to be affected, because many antibiotics are also metabolized by the CYP450 system. For example, clarithromycin, a macrolide antibiotic used for Helicobacter pylori treatment, is a potent CYP3A inhibitor. The coadministration of TAC and clarithromycin may increase serum TAC levels four- to five-fold (Fig. 95–5).89 Another example is the use of fluoroquinolones such as ciprofloxacin, norfloxacin, or levofloxacin, which are also metabolized by the P450 system. A small study tested whether levofloxacin, an agent with limited hepatic metabolism, affected blood levels of CSA or TAC. Levofloxacin significantly increased the mean area under the blood concentration-time curve (AUC) of CSA and TAC by approximately 25%.90
Figure 95–5.
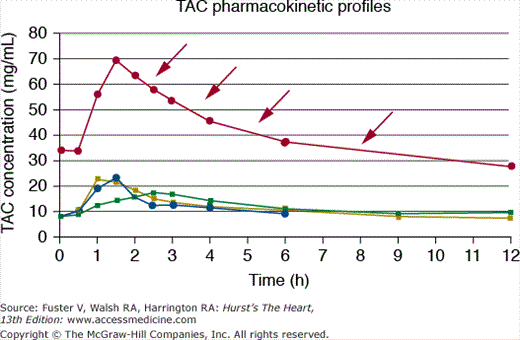
Significant interaction between tacrolimus (TAC) and clarithromycin (CLA). Pharmacokinetic profiles are shown for various TAC doses before CLA (8 mg/dand 6 mg/d), with CLA (4 mg/d, filled circle), and 2 months after CLA (4 mg/d, filled square). Note the marked increase in serum TAC levels in the presence of CLA despite lower TAC dosing (arrows). Reproduced with permission from Kunicki PK, Sobieszczan`ska-Malek M. Pharmacokinetic interaction between tacrolimus and clarithromycin in a heart transplant patient. Ther Drug Monit. 2005;29(1):107-108.
See Table 95–2 for a complete list.
The target of rapamycin (TOR) inhibitors have become more commonly used in heart transplant recipients. Sirolimus (SIR) was the first introduced in the market, and now everolimus (EVER) has been approved for use in cardiac transplant patients. Both of these agents are macrolide immunosuppressants. SIR is extensively metabolized by CYP3A4, and therefore drug interactions are likely. Both of these agents exacerbate hyperlipidemia, and therefore statin use requires the same precautions as with the CYP3A inhibitors.84
In current posttransplant practice, it is common to use combinations of agents with the CYP3A inhibitor drugs. The administration time of CSA with SIR may affect SIR pharmacokinetics. There are data that support increases in SIR levels depending on the time of administration of CSA. When CSA and SIR are administered together, SIR levels increase, possibly due to inhibition of first-pass metabolism. Therefore SIR should be administered 4 hours after CSA dosing.91
The azole-derived antifungal agents should be used carefully in combination with SIR or EVER. See Table 95–3.
| Drug | Effect | Onset | Management |
|---|---|---|---|
| Diltiazem | Increased SIR effect | Delayed | Monitor SIR levels 3 times per week in first week |
| Fluconazole | Increased SIR/EVER effect | Delayed | Monitor SIR/EVER levels for 1-2 wk |
| Itraconazole | Increased SIR/EVER effect | Delayed | Monitor SIR/EVER levels for 1-2 wk |
| Ketoconazole | Increased SIR/EVER effect | Delayed | Avoid combination |
| Voriconazole | Increased SIR/EVER effect | Delayed | Avoid combination |
| Cyclosporine | Increased SIR/EVER effect | Rapid | Administer SIR 4 h after cyclosporine |
Mycophenolate mofetil (MMF) is an antiproliferative drug that is well absorbed after oral administration and converts to its active metabolite mycophenolic acid (MPA). MPA is metabolized by glucuronyl transferase and excreted in the urine and bile. When cyclosporine and MMF are given in combination, the result may be lower plasma MMF levels secondary to cyclosporine-induced alterations in biliary clearance. The effects of concurrent tacrolimus administration on MPA exposure are less clear.92
Cholestyramine may decrease MMF active compound levels. This decrease is probably due to binding of the recirculating conjugated active compound by cholestyramine, preventing enterohepatic circulation of MMF and loss of the secondary peak.93 Package labeling recommends that MMF and cholestyramine not be coadministered.
The absorption of MMF may be impaired by antacids or iron preparations because of possible chelation complex formation. Therefore, it is advisable to stagger any antacids or iron supplements 2 to 4 hours with MMF administration.
Azathioprine is not as widely used today as it was in the 1990s as an antiproliferative agent. The reader is referred to several in-depth discussions of the pharmacokinetics of azathioprine and its potential ADRs.94,95
Coronary Artery Disease
Major technologic and pharmacologic advances over the past 2 decades have substantially improved outcomes in patients with acute and chronic coronary artery disease. The widespread utilization of fibrinolytic, antiplatelet, and catheter-based therapies has dramatically improved the morbidity and mortality of coronary disease, leveling its multiple manifestations (see Chap. 61). This changing clinical landscape increases the potential for adverse drug interactions, and knowledge of this potential is mandatory to provide safe, effective care to patients with coronary artery disease.
Although urgent catheter-based coronary revascularization has been shown to be superior to fibrinolytic therapy in patients presenting with ST-segment elevation myocardial infarction (STEMI), lack of timely proximity to interventional cardiology services often precludes this treatment option (see Chap. 63). Fibrinolysis thus remains an essential component to STEMI management for many patients. All fibrinolytic drugs work by either directly or indirectly promoting the conversion of plasminogen to plasmin, a nonspecific serum protease that lyses fibrin clot and degrades certain clotting factors.96 The risk for potentially fatal bleeding complications with the use of any fibrinolytic agent is self-evident, and full knowledge of the absolute and relative contraindications of fibrinolytic drugs is mandatory before their use.
The risk for significant pharmacokinetic interactions involving fibrinolytic drugs is low. Fibrinolytics are not dependent on cytochrome P450 metabolism and not much affected by inhibitors and inducers of this enzyme.96 Agents such as alteplase and saruplase are highly cleared by the liver and thus are theoretically susceptible to reduced hepatic blood flow, which might occur in patients receiving β-blockers, nitrates, or with cardiogenic shock. There are a few potential pharmacodynamic interactions to consider when using fibrinolytic drugs. Concomitant use of heparin does increase the potential for serious bleeding in patients treated with fibrinolytic agents; however, in general this increased risk does not offset the additive benefit of these drugs with respect to maintaining vessel patency97,98 and reducing recurrent ischemic events, including recurrent MI. The activated partial thromboplastin time (aPTT) should be frequently monitored when heparin is used in conjunction with a fibrinolytic agent and should be maintained between 1.5 and 2.0 times the upper limit of normal.99 Aspirin does not appear to increase the risk of bleeding when given with fibrinolytic therapy and in fact improves mortality. Data from clinical trials suggest that glycoprotein (Gp) IIb/IIIa inhibitors may result in an unacceptably high risk for bleeding when given with full-dose fibrinolytic therapy, and protocols using half-dose fibrinolytics, while promising on measures of vessel patency, have been uniformly disappointing in failing to improve clinical outcomes.100
Aggressive platelet inhibition has revolutionized the medical and percutaneous management of coronary artery disease. Indeed, the broad success and use of intracoronary stenting is due in part to the development of potent antiplatelet agents used alone and in combinations that prevent catastrophic early and late stent thrombosis and improve long-term clinical outcomes.
Aspirin plays a cornerstone role in the secondary prevention of coronary artery disease, and its use is essential, along with an adenosine diphosphate (ADP) blocker, such as clopidogrel or prasugrel, to reduce the risk of intracoronary stent thrombosis. Like other inhibitors of cyclooxygenase-1, aspirin reduces prostaglandin production, which may attenuate the effects of many antihypertensive drugs, though this phenomenon is more likely to occur at higher (>100 mg) aspirin doses. Aspirin hypersensitivity, though rare, may result in life-threatening bronchospasm and anaphylaxis. Patients with true aspirin hypersensitivity who have a strong indication for the drug may undergo rapid desensitization within a few hours, preferably under the consultative care of an allergist expert in the procedure.101 Aspirin use in conjunction with anticoagulants can increase the likelihood for significant bleeding complications.102
Ticagrelor is a new platelet aggregation inhibitor that was associated with a reduction in the composite of death from vascular causes, myocardial infarction, or stroke when compared with clopidogrel in patients with acute coronary syndromes.103 There was no difference in the overall rate of major bleeding, but the ticagrelor group did have a higher rate of non–coronary artery bypass grafting–related bleeding. Ticagrelor blocks ADP receptors of subtype P2Y12, but the inhibition is reversible. Unlike the ADP blockers clopidogrel and prasugrel, ticagrelor is not a pro-drug and does not require hepatic activation. It does have some other adverse effects, including an increased incidence of bradycardia and dyspnea compared with clopidogrel. Ticagrelor is being evaluated by the FDA for approval in clinical use.
The thienopyridines (ticlopidine and clopidogrel) inhibit platelet function by binding to platelet surface ADP receptors. Both ticlopidine and clopidogrel, alone or in combination with aspirin, have been shown to reduce the likelihood of recurrent myocardial ischemia or infarction in at-risk populations, and thienopyridine therapy is essential after intracoronary stenting.102,104
Ticlopidine is a potent inhibitor of CYP2D6 and CYP2C19 and thus carries a risk for pharmacokinetic interactions with drugs metabolized by these enzymes. Ticlopidine use has been associated with adverse hematologic events, including aplastic anemia and thrombotic thrombocytopenic purpura.105 Since the development of clopidogrel, the use of ticlopidine has been limited largely to patients who are clopidogrel intolerant.
Clopidogrel is associated with fewer adverse reactions than ticlopidine and is administered once daily. Hematologic abnormalities associated with clopidogrel use are rare and rashes are uncommon.106 Clopidogrel is activated by CYP3A4, and its antiplatelet effects may be lessened by concurrent use of CYP3A4 promoters such as amiodarone. Pharmacodynamically, clopidogrel use is associated with an increased risk for significant bleeding when given with anticoagulants or other antiplatelet agents, including aspirin. The relevance of this interaction depends on the clinical entity being treated. In patients with acute coronary syndromes, the combination of aspirin and clopidogrel has been shown to significantly reduce recurrent adverse cardiac events to a greater degree than it promotes major bleeding.107,108 Thus, in this population, dual antiplatelet therapy is preferred. The same cannot be said for patients presenting with stroke. Here, aspirin and clopidogrel may reduce the risk for recurrent events when given individually, but in combination, these drugs do not incrementally reduce event rates enough to justify their increased combined bleeding risk.109
Some proton pump inhibitors may inhibit the metabolism of clopidogrel to its active metabolite through inhibition of CYP P-450-2C19. With this mechanism as a background, some retrospective studies have suggested an increase in cardiovascular events in patients treated with proton pump inhibitors and clopidogrel.110-112 Although there does appear to be both a pharmacokinetic and pharmacodynamic interaction when combining these drugs, other observational data have been less compelling regarding a true effect (or interference) on clinical outcomes.113 On November 17, 2009, The FDA issued a Health Care Provider alert making clinicians aware of these potential interactions and urging caution in prescribing the drugs concomitantly (http://proxy.library.upenn.edu:2540). Definitive data to better guide practice are needed from an appropriately designed randomized clinical trial.
Prasugrel is a new member of the thienopyridine class of ADP receptor inhibitors that inhibits platelet aggregation by irreversibly binding to P2Y12 receptor. In patients with acute coronary syndrome (TRITON-TIMI 38), prasugrel (60-mg loading dose followed by 10-mg daily dose) reduced the combined rate of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke compared with clopidogrel.114 The difference was primarily driven by the reduction of nonfatal myocardial infarctions. This improvement in outcomes was associated with increased rate of serious bleeding events and fatal bleeding with prasugrel. Overall mortality did not differ between the groups. In July 2009, the FDA approved prasugrel as the new antiplatelet drug while issuing a black box warning about its increased bleeding risk and urging caution in considering its use in certain patient groups. Prasugrel is contraindicated in patients with prior history of TIA or stroke and generally is not recommended in patients older than 75 years. Additional risk factors for bleeding with prasugrel include weight less than 60 kg and use with other medications that increase the risk of bleeding. Dose reduction to 5-mg daily dose should be considered in patients less than 60 kg, although the data supporting this reduced dosing recommendation are largely based on pharmacokinetic and pharmacodynamic considerations rather than on clinical outcomes.
Dipyridamole is a potent vasodilator with antiplatelet activity that is often used as the vasodilator in pharmacologic myocardial perfusion imaging studies. It also has a role as an adjunctive antiplatelet agent in certain patients with cerebrovascular disease; its role as an effective agent added to other antiplatelet agents in patients with coronary artery disease has not been established. The vasodilatory effects of dipyridamole are antagonized by xanthine derivatives, and adverse reactions to dipyridamole such as bronchospasm, bradycardia, and flushing can be treated with theophylline or aminophylline.
Therapeutic anticoagulation with heparinoids and direct thrombin inhibitors has dramatically improved outcomes in patients with coronary artery disease, particularly acute coronary syndromes. The risk for potentially serious bleeding with antithrombin therapy is obvious; however, with appropriate monitoring, the benefit these drugs provide to patients with unstable coronary syndromes far outweighs their collective risk.
Heparin potentiates the effect of antithrombin III, which leads to inactivation of thrombin. Heparin also inactivates several clotting factors and prevents the conversion of fibrinogen to fibrin. Heparin has a half-life of approximately 90 minutes and is metabolized by the liver and reticuloendothelial system. Pharmacokinetic drug interactions involving heparin are rare, and most pharmacodynamic interactions with heparin involve the concurrent use of drugs with antiplatelet or anticoagulant properties such as aspirin or warfarin. Life-threatening bleeding complications involving heparin can be treated with its antidote, protamine sulfate. Early, abrupt cessation of heparin therapy in patients treated for acute coronary syndromes has been associated with rebound ischemia.115 Monitoring these individuals for at least 24 hours after heparin cessation is advisable.
One potentially dangerous adverse event associated with heparin use is heparin-induced thrombocytopenia (HIT). Mild thrombocytopenia may occur in as many as 20% of patients beginning heparin therapy and typically resolves within a few days. HIT occurs in 1% to 5% of heparin-exposed patients and is associated with significant thrombocytopenia (<100,000/μL) that typically occurs several days after exposure to any amount of heparin, although cases of delayed-onset HIT have been described.116 HIT is caused by autoantibodies directed against the complex of heparin and platelet factor 4 (PF4).117 These antibodies may trigger platelet activation and produce a prothrombotic state that places affected patients at risk for venous and arterial thrombosis. HIT management includes discontinuation of heparin and in some cases anticoagulation with direct thrombin inhibitors and warfarin. A prior history of HIT is considered a contraindication to subsequent heparin therapy.
Low molecular weight heparin (LMWH) is produced by chemical or enzymatic depolymerization of the unfractionated heparin molecule. This process produces small (4000-6500 Da) molecules that maintain activity against factor Xa with less potential to interact with other molecules including platelet factor 4.118 The LMWH enoxaparin has established efficacy in the management of patients with acute coronary syndromes modestly superior to that of heparin, with the advantage of subcutaneous administration and fixed dosing that does not require adjustment or serial monitoring.119,120 LMWH is renally excreted and its use in patients with severe kidney disease is relatively contraindicated. Like heparin, LMWH is not associated with a high risk for pharmacokinetic drug interactions. The potential for pharmacodynamic interactions with antiplatelet and other anticoagulant drugs is understood, and in instances where bleeding is severe, the effects of LMWH can be partially reversed by protamine sulfate. LMWH use is associated with a lower risk (but not a zero risk) for HIT compared with unfractionated heparin. However, because LMWH has been shown to cross-react with HIT antibodies in up to 70% of patients with known HIT, the use of LMWH as a heparin alternative in patients with HIT is not advised especially considering the availability of the direct thrombin inhibitors.117,121,122
The direct thrombin inhibitor bivalirudin has established efficacy as an alternative to heparin in patients with acute coronary syndromes who require percutaneous revascularization, particularly in the setting of renal insufficiency.123 Bivalirudin binds directly to thrombin at its catalytic site and reversibly inhibits circulating and clot-bound thrombin.124 The drug is proteolytically cleaved and excreted in the urine; its anticoagulation effects resolve within an hour of discontinuation. Bivalirudin is not associated with significant pharmacokinetic interactions. The potential for pharmacodynamic interactions when given with other anticoagulant and antiplatelet agents exists, but in general, bivalirudin use is associated with a lower risk for significant bleeding than heparin.
The majority of patients presenting with acute coronary syndromes will benefit from early invasive management of their disease, and the catheterization laboratory is often the site of first exposure to potentially hazardous pharmacologic agents.125
Iodinated contrast agents are associated with several potential adverse reactions. Hypersensitivity reactions occur in approximately 1% of patients treated in the catheterization laboratory and can range in severity from a mild rash to airway compromise and hemodynamic collapse. Most severe contrast reactions are anaphylactoid (non–immunoglobulin E mediated) reactions that involve the release of molecules such as histamines and leukotrienes from mast cells and tend to occur within minutes of contrast exposure.126 Pretreatment of patients with a known contrast allergy with oral corticosteroids at least 6 hours before catheterization and antihistamines just before catheterization may reduce the likelihood of contrast reactions.127 The efficacy of intravenous corticosteroid administration just before catheterization has not been established, though it may make the operator feel better. Acute management of hemodynamically unstable patients or those with airway compromise may include intravenous epinephrine or methylene blue.128,129 Although the use of protamine sulfate during catheterization is now rather uncommon, hypersensitivity reactions may occur with administration of this heparin antidote, particularly in patients treated with NPH insulin. Contrast agents may also produce contrast-induced nephropathy (CIN), which may lead to permanent kidney damage or dialysis in a minority of patients. Underlying kidney disease appears to be the greatest risk factor for CIN, and prophylactic hydration and treatment with N-acetylcysteine may have a modest effect on reducing the risk for CIN, although the clinical outcome data are not definitive.130 Lastly, patients taking metformin for diabetes are at risk for rare but potentially lethal lactic acidosis. Metformin-related lactic acidosis tends to occur most commonly in patients who develop CIN after contrast administration. It is advisable to withhold metformin for 24 to 48 hours after catheterization because this is the period when CIN typically occurs.131,132
The introduction of sirolimus- and paclitaxel-coated coronary stents has significantly reduced in-stent restenosis rates, and drug-eluting stents (DES) have grown to dominate the market since their debut.133 There appear to be significant differences between these stents with respect to their drug-release kinetics and influence on cell line proliferation that may have clinical relevance.134 There is also growing concern over reports of late stent thrombosis with DES, particularly after antiplatelet agent withdrawal.135-137 Although rare, local and systemic hypersensitivity reactions involving DES have been reported and may contribute to late stent thrombosis risk.138 These factors may result in prolongation of dual antiplatelet therapy after DES deployment and raise concerns about the safe management of patients who require discontinuation of antiplatelet therapy due to bleeding or a compelling indication for surgery. (See Chap. 61 for more information.)
Rhythm Disorders
Despite the recent attention given to nonpharmacologic rhythm management options such as device therapy and catheter-based ablation procedures, many patients with rhythm disorders (ventricular as well as supraventricular) will require some form of antiarrhythmic therapy.139,140 The potential for significant adverse drug interactions involving antiarrhythmic drugs is enormous, for reasons related not only to the drugs themselves but also to comorbidities of the patients taking them. Antiarrhythmic drugs in general function within a narrow therapeutic spectrum; minor alterations in serum levels may result in loss of efficacy or overt toxicity. Many of these agents are dependent on oxidative metabolism via subtypes of cytochrome P450, thus allowing for possible interactions with the ever-growing list of inducers and inhibitors of these enzymes. Oxidative metabolism can also vary due to patient-specific factors. Polymorphisms involving the genes that code for P450 subtypes such as CYP2D6 and CYP2C19 have recently been described and appear to vary with ethnicity.141 Additionally, patients with rhythm disorders often have structural heart disease and comorbid conditions that require their own cadre of medications, further increasing the potential for concerning adverse events. Avoiding adverse drug interactions in patients with rhythm disorders can be challenging. This section focuses on the more common or concerning interactions of specific antiarrhythmic drugs, organized by the Vaughn Williams classification scheme.142



