Introduction: prevalence of congenital heart disease
Congenital heart disease (CHD) is defined as a cardiovascular abnormality that is present from birth, and approximately 4–10 liveborn infants per 1000 are affected.1,2 However, the true prevalence may be much higher, as these estimates often exclude bicuspid aortic valve and prolapse of the mitral valve, the most common cardiac malformations.1 A more accurate estimation has been reported to be around 50 per 1000 live births.1 Table 63.1 summarizes the recently reported incidence of the common congenital heart defects.2 The great successes of pediatric cardiac care in the past 25 years have resulted in a marked increase in adult patients with CHD. About 85% of children with CHD are now expected to survive into adulthood.3 The most recent epidemiologic study, conducted in Quebec, Canada, revealed that the prevalence of adults with CHD was 4.09 per 1000 for the year 2000, representing an increase of 85% compared with 1985, while this increase was only 22% in children for the same period.4 The authors extrapolated their results to the US population and estimated that about 900 000 adults had CHD in the year 2000 in the USA.
Table 63.1 Incidence per 10 000 live births
| Lesion | Median |
Bicuspid aortic valve (BAV) | 92.4 |
Ventricular septal defect (VSD) | 28.3 |
Patent ductus arteriosus (PDA) | 5.7 |
Atrial septal defect (ASD) | 5.6 |
Pulmonary stenosis (PS) | 5.3 |
Aortic coarctation | 3.6 |
Tetralogy of Fallot (TOF) | 3.6 |
Atrioventricular septal defect (AVSD) | 3.4 |
Complete transposition of the great arteries (d-TGA) | 3.0 |
Aortic valve stenosis (AS) | 2.6 |
Modified with permission from Hoffman and Kaplan.2
Most early interventions carried out on patients with CHD have not been curative and a significant proportion of them will need further surgery or experience various complications such as arrhythmia and heart failure. Their cardiovascular disease is complex and their management poses a real challenge, requiring expert cardiologic care. Indeed, initial assessment of suspected CHD, follow-up of patients with CHD of moderate severity and complex lesions as well as risk assessment for non-cardiac surgery and pregnancy should ideally take place in a tertiary adult care center. However, there are currently not enough such specialists worldwide. Education on the principles of CHD for a broader professional audience is therefore imperative to improve the lifetime care of this population.5 Primary care physicians, general adult cardiologists, obstetricians, surgeons and anesthesiologists should also be aware of the various issues in the general medical management of CHD because most patients will also need local follow-up for economic, social and geographic reasons.6,7
This chapter provides key clinical information and guidelines for the management of adults with CHD.7a
Genetic basis of CHD
In recent years, there have been tremendous advances not only in diagnosis and treatment of CHD but also in our knowledge of the etiologies of CHD. Improvements in genetic techniques have resulted in identification of several loci and genes associated with CHD. In their extensive review of the current genetic basis for congenital heart defects, Pierpont et al1 emphasized the reasons why it is very important to determine whether there is an underlying genetic abnormality:
- other organ systems may be involved
- it may provide prognostic information
- there may be significant risks of transmission to the offspring
- genetic testing in other family members may be appropriate.
A chromosome deletion can be identified by fluorescence in situ hybridization (FISH) in some syndromes associated with heart defects. For example, a 22q11 deletion is present in ∼90% of patients with DiGeorge syndrome, which is characterized by cardiac defects (such as aortic arch anomalies, truncus arteriosus, tetralogy of Fallot (TOF) or ventricular septal defect (VSD)), specific facial features, palate anomalies, hypoplasia of the thymus, aplasia or hypoplasia of the parathyroid glands, and psychiatric disorders. The 22q11 deletion syndrome is an autosomal dominant syndrome which carries a 50% risk of transmission from an affected parent to the offspring.1 Therefore, FISH testing should be considered for patients with such cardiac defects, especially if one of the other features of the 22q11 deletion syndrome listed above is present.1 A chromosome microdeletion (7q11.23) is also found in ∼ 90% of patients with Williams syndrome, which is associated with supravalvular aortic stenosis (AS) and often supravalvular or peripheral pulmonary stenosis (PS). Mutations in various single genes have been found in several syndromes, namely Holt–Oram, Alagille, Char, Noonan, LEOPARD, CHARGE association, Ellis –van Creveld and Marfan. Single-gene disorders can also result in non-syndromic congenital heart defects, such as familial congenital atrial septal defect (ASD) and atrioventricular block (AVB).1 However, a more complex combination of multiple genetic alterations and environmental factors instead of a single gene mutation is likely to be the cause in many cases of non-syndromic CHD.1 Our knowledge of the association between CHD and genetic alterations will continue to grow with ongoing research and current availability of genetic testing.
Non-inherited risk factors for CHD
During the past decade, there has been an increase in epidemiologic literature concerning modifiable risk factors that may have an adverse effect on the fetal heart,8 especially with an exposure during the first pregnancy trimester and the 3 months before conception (Table 63.2).8
We now provide information on specific lesions.
Table 63.2 Definite or possible risk factors associated with CHD in offspring
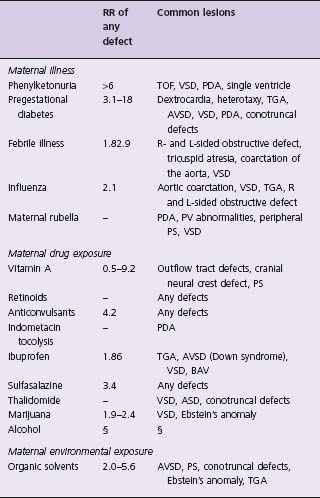
Modified with permission from Jenkins et al.8
– Not available.
§ Inconclusive data on CHD.
R, Right; L, Left; PV, pulmonary valve.
Anatomic findings and pathophysiology
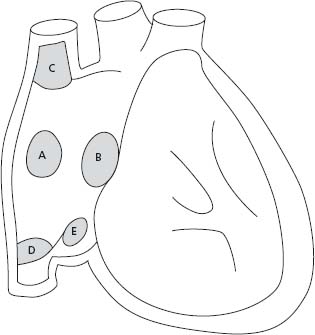
Interatrial communications are common and can be classified in four types: ostium secundum defects, the most common, are located within the region of the fossa ovalis; ostium primum, part of an atrioventricular septal defect (AVSD), includes a common atrioventricular junction with a trileaflet left atrioventricular valve (AVV); superior sinus venosus defects are frequently associated with partial anomalous pulmonary vein return; and coronary sinus defects are characterized by a deficiency in the roof between the coronary sinus and the left atrium (Fig. 63.1). The size of the defect and the ventricular diastolic filling pressures (right and left) determine the degree of left-to-right shunting. A Qp: Qs ratio greater than 1.5/1.0 or the presence of right heart dilation is considered an indication for closure.9
Figure 63.1 Anatomic location of ASDs. A, indicates secundum ASD; B, ostium primum ASD or partial AV septal defect; C, superior sinus venosus ASD; D, inferior sinus venosus ASD; E, coronary sinus ASD.
Clinical presentation
Dyspnea and fatigue on exertion are the most common symptoms in adults. Patients can also present with palpitations from atrial fibrillation or flutter, which rarely occurs before 40 years of age.10,11 On examination, a right ventricular lift and a wide and fixed splitting of the second heart sound are distinctive clinical signs of ASD. A pulmonary ejection systolic murmur and a mid-diastolic rumble reflect increased flow through the pulmonary and tricuspid valves.
Diagnostic evaluation
Electrocardiogram
The ECG in ASD can reveal sinus rhythm, atrial fibrillation or atrial flutter, rightward axis deviation in secundum ASD (leftward or extreme right in primum ASD), prolonged PR interval, and right bundle branch block pattern (RBBB).12–14 In superior sinus venosus defect, sinus node dysfunction may lead to inverted P-waves in inferior leads.10,15,16
Chest radiography
Signs of increased pulmonary blood flow,17,18 as well as enlarged central pulmonary arteries, cardiomegaly (right heart dilation), and small aortic knuckle from chronic low systemic cardiac output, are present in significant defects.10
Exercise testing
Useful to document exercise capacity when there is discrepancy between symptoms and clinical findings, or to show changes in oxygen saturation in patients with mild or moderate pulmonary arterial hypertension (PAH) (Class IIa, Level C).
Echocardiography
Transthoracic echocardiogram (TTE) usually demonstrates the location, shape and size of the defect and its septal margins.19 TTE can also provide the direction of the shunt, an estimation of the shunt ratio (Qp:Qs)20 and an estimation of the pulmonary artery pressures (PAP) from the Doppler velocity of the tricuspid regurgitation (TR). An enlarged right ventricle (RV),21 with or without paradoxic septal motion.22 might be the only initial findings of a significant ASD. Transesophageal echocardiogram may better visualize the pulmonary venous return, and is used to guide device closure of the defect.
Cardiac magnetic resonance imaging (CMR)
Cardiac magnetic resonance imaging is the modality of choice to assess right ventricular size and function. Pulmonary venous return can also be accurately demonstrated.23
Management
Indications for ASD closure are shown in Table 63.3.7a,10 Most centers favor percutaneous closure when the anatomy is suitable.24,25 Device closure is safe (complication rate less than 1%) and effective, and minimizes hospital stay.26 It also improves RV size and function27 and exercise capacity,24 as does surgical closure. However, surgical closure is required for patients with ostium primum and sinus venosus ASDs (Class I, Level B), as well as ostium secundum defects with one or more of the following features: stretched diameter >36–40 mm, inadequate atrial septal rims to allow device deployment, or proximity of the defect to the AVV, coronary sinus or vena cavae10,25 (Class IIa, Level C).
Table 63.3 ASD closure
Indications ASD associated with RA and RV enlargement, with or without symptoms (Class I, Level B) Paradoxic embolism (Class IIa, Level C) Documented orthodeoxia-platypnea (Class IIa, Level B) Contraindications Advanced pulmonary arterial hypertension (PAH) (Class III, Level B) Pregnancy (defer closure for 6 months after delivery) (Level C) Severe left ventricular dysfunction (Level C) |
RA, Right atrium.
Outcomes and complications
Normal long-term survival has been reported after surgical ASD closure when patients were operated on before 25 years of age.28 Nevertheless, benefits (reduced morbidity and mortality) have also been demonstrated with surgical ASD closure compared with medical treatment in patients over 40 years of age.29,30 The risk of developing late atrial flutter or fibrillation is clearly higher if surgical repair is performed after 40 years of age.11 Other complications are summarized in Table 63.4.
Table 63.4 Complications of ASD
Arrhythmias11,28 Atrial flutter or fibrillation Bradyarrhythmia Impaired functional capacity31,32 Right heart failure PAH (if large, unrestrictive defect) Cerebrovascular events from paradoxic embolism (uncommon) |
Anatomic findings and pathophysiology
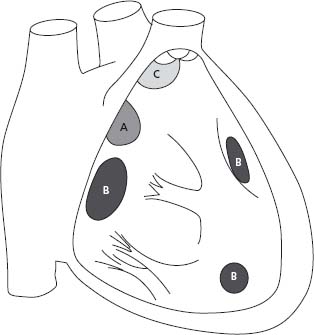
Ventricular septal defect (VSD) is a very common congenital heart malformation (up to 53.2 per 1000 liveborn infants).33 VSDs are classified into (1) perimembranous, (2) muscular, and (3) double committed subarterial (Fig. 63.2). In perimembranous VSDs, part of the rim of the defect is formed by part of the central fibrous body, which is between the leaflets of an AVV and an arterial valve. Muscular VSDs possess completely muscular rims. Finally, double committed subarterial VSDs are present when the aortic and pulmonary valves are contiguous in the roof of the defect.34
Figure 63.2 Anatomic location of VSDs. A, indicates perimembranous VSD; B, muscular VSDs; C, doubly committed and subarterial VSD.
The magnitude of and direction of flow through the VSD are determined by the size of the defect, the right and left ventricular pressures, and the pulmonary vascular resistance. In small (restrictive) VSDs, there is a significant pressure gradient between the left and right ventricles. In large non-restrictive VSDs, the pulmonary pressures are at or close to systemic levels. With the development of pulmonary vascular disease, the degree of left-to-right shunting decreases and then reverses, leading to Eisenmenger syndrome.
Clinical presentation
Patients with small VSDs are usually asymptomatic. Moderately restrictive and large VSDs can cause dyspnea on exertion, and/or palpitations if arrhythmias arise. As mentioned above, non-restrictive VSDs can lead to Eisenmenger syndrome. On examination, a holosystolic murmur is heard on the left sternal edge, which is loudest and may be accompanied by a thrill in small defects. In moderate and large defects, the apical impulse is laterally displaced. There is a widely split second heart sound that varies with respiration. With large shunts, a diastolic rumble of increased mitral flow may be heard. Large non-restrictive VSDs may have signs of PAH, including a RV heave and a loud P2.
Diagnostic e valuation
Electrocardiogram
In small VSDs, the ECG is usually normal. In moderate VSDs, ECG may reveal left atrial hypertrophy and signs of left ventricular overload. In large non-restrictive VSDs with PAH, the QRS axis shifts to the right and right atrial/ ventricular hypertrophy is present. Following surgical repair, RBBB is found in 29–65% of patients more than 25 years after surgery35,36 and 0.7–3.5% may develop late complete AVB.36,37
Chest radiography
Small VSDs have normal chest X-rays. Pulmonary plethora and cardiomegaly are present with larger defects. When significant PAH develops, the chest X-ray shows dilated central pulmonary arteries and right heart enlargement.
Echocardiography
The size and location of VSDs can be accurately assessed by TTE.38,39 Shunt ratio (Qp:Qs) and PAP can be estimated20 and, most importantly, hemodynamic consequences such as left ventricular enlargement or hypertrophy can be ascertained. Associated lesions should be sought (aortic regurgitation (AR), right or left ventricular outflow tract obstruction). Transesophageal or three-dimensional echocardiography may be occasionally required when the TTE quality is poor.40
Cardiac catheterization
Cardiac catheterization is useful when non-invasive data are inconclusive and further information is needed (Class IIa), such as assessment of pulmonary vascular resistance, magnitude of shunts, and pulmonary vasoreactivity (Level B).
Management
Table 63.5 summarizes the indications for VSD closure.7a Surgical closure in the current era carries low perioperative mortality risk (less than 1%),41 with a low risk of AVB requiring permanent pacemaker (0.7–3%).37,41,42 Transcatheter closure is currently in the investigational stage and should be considered only in selected cases of muscular and perimembranous VSDs (Class IIb, Level C). Butera et al recently reported their 38.5 months follow-up of 104 patients who underwent percutaneous closure of perimembranous VSDs, and the occlusion rate reached 99% at follow-up. However, pacemaker implantation for complete AVB occurred in 5.6% (all in patients <6 years old).
Table 63.5 VSD closure
Indications Left-to-right shunt (Qp: Qs) >2 by echocardiography, CMR or cardiac catheterization and evidence of LV volume overload (Class I, Level B) Left-to-right shunt (Qp: Qs) >1.5: 1 with pulmonary artery pressure < two-thirds of systemic pressure and PVR < two-thirds of systemic vascular resistance (Class IIa, Level B) Left-to-right shunt (Qp: Qs) >1.5: 1 in the presence of LV systolic or diastolic dysfunction (Class IIa, Level B) Regardless of the size: History of endocarditis (Class I, Level C) May be considered in patients with aortic regurgitation (Level C) Contraindications Advanced PAH (Class III, Level B) Pregnancy (defer closure for 6 months after delivery) (Level C) |
LV, left ventricle; E F, ejection fraction.
Outcomes and complications
Small restrictive VSDs with normal PVR have an excellent prognosis.43 Twenty-five year survival rate decreases with increasing size of the VSD (small, 96%; moderate, 86%; large, 61%; Eisenmenger, 42%).44 Following surgical closure in patients without previous PAH, life expectancy should be close to normal. Even small VSDs, in unoperated patients or as residual defects following surgery, can lead to complications such as endocarditis, aortic regurgitation or arrhythmias.45,46 Other possible complications of VSDs are listed in Table 63.6.
Table 63.6 Complications of VSDs
Arrhythmias47 Atrial fibrillation Ventricular arrhythmias, sudden cardiac death Aortic regurgitation Endocarditis LV dysfunction (in large defects) PAH and Eisenmenger syndrome (if large, unrestrictive defect) |
Atrioventricular septal defect
Anatomic findings and pathophysiology
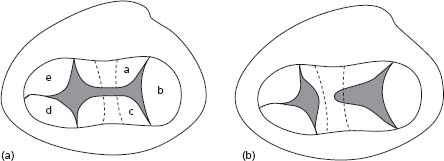
The morphologic hallmark of an AVSD is a common atrioventricular junction, with abnormalities of the AVV. In complete AVSD, there is a common AVV with five leaflets, while in partial AVSD (ostium primum ASD), the AVV is divided into two orifices (Fig. 63.3). The left AVV has three leaflets (mural leaflet, inferior and superior bridging leaflets). In complete AVSD, there is usually a primum ASD and a large VSD. In partial AVSD, only a primum ASD is present. Other common features include an inlet-to-outlet disproportion of the left ventricle and unwedged position of the aortic valve, which result in an elongated left ventricular outflow tract (LVOT) (“goose neck deformity”), predisposing to subaortic obstruction. Septal chordal attachments of the left AVV contribute to this complication.48 The conduction system is also abnormal in patients with AVSD, leading to an increased risk of AVB.49
Figure 63.3 Complete and partial AVSDs. A illustrates common valvar orifice (a, superior bridging leaflet; b, left mural leaflet; c, inferior bridging leaflet; d, right inferior leaflet; e, right antero-superior leaflet). B illustrates separate right and left valvar orifices.
Complete AVSD is associated with abnormal karyotype in 49% of cases diagnosed in fetal life (39% having trisomy 21),51 while some surgical series report Down syndrome in up to 73% of patients undergoing complete AVSD repair.52
Complete AVSDs have a significant degree of left-to-right shunting through the primum ASD and the large VSD, leading to left ventricular enlargement and early PAH. Partial AVSDs (with only a primum ASD) will usually have similar pathophysiology to other isolated interatrial shunts, unless the left AVV is very dysplastic, leading to severe stenosis and/or regurgitation.
Clinical presentation
The clinical presentation relates to the morphology of the defect. Large left-to-right shunts and severe left AVV regurgitation will present with congestive heart failure (CHF) in infancy and will develop PAH and cyanosis. Adults with unrepaired complete AVSDs may present with exercise intolerance and fatigue, Eisenmenger syndrome (see below), palpitations from atrial arrhythmias or bradycardia from complete AVB. Examination may reveal cyanosis and clubbing, right ventricular impulse, increased pulmonary component of the second sound, variable ejection systolic murmurs, mid-diastolic murmur in large left-to-right shunt, and pansystolic murmur with left AVV regurgitation. Isolated ostium primum ASDs will share the clinical features of ASDs described above but may also have signs of left AVV dysfunction.
Diagnostic evaluation
Electrocardiogram
Left axis deviation, RBBB, and first-degree AVB are common ECG findings in patients with AVSD.53,54 Atrial fibrillation can also be present.53
Chest radiography
Cardiomegaly from left heart dilation usually present in patients with large VSD and/or significant left AVV regurgitation. Large primum ASD will give rise to right heart enlargement and increased pulmonary vascular markings. Patients with Eisenmenger syndrome will have dilated central pulmonary arteries and peripheral pruning.
Echocardiography
Diagnosis of AVSD can accurately be made by TTE.55 The following features should be assessed.56–58
- Abnormal configuration of the AVVs are at the same level.
- Left AVV has three leaflets.
- May have some degree of regurgitation.
- Presence of ASD and VSD, and the magnitude of left-to-right shunting.
- Estimated right ventricular systolic pressure.
- Inlet-to-outlet disproportion with elongated LVOT and possible LVOT obstruction.
- Abnormal position of the papillary muscles.
- Left ventricular size and function.
Cardiac magnetic resonance
This provides detailed morphologic information and accurately estimates the size of the VSD.59
Cardiac catheterization and angiography
Echocardiography and CMR have replaced cardiac catheterization for the diagnostic and preoperative evaluation of AVSDs. However, cardiac catheterization may be necessary to assess formally the pulmonary vascular resistance and vasoreactivity (Class IIa, Level B).
Management
Patients with complete AVSD should have early surgical correction (including repair of the AVV60,61) within the first few months of life to prevent pulmonary vascular disease. Most patients who present with partial AVSDs in later life (>40 years old) will also benefit from surgical repair.62 However, patients with irreversible pulmonary vascular disease and/or Eisenmenger syndrome should usually be treated medically (see below). Indications of reintervention in adults include:7a
- significant left AVV regurgitation or stenosis with symptoms, atrial or ventricular arrhythmias, a progressive increase in LV dimensions or deterioration of LV function (Class I, Level B)
- significant LVOT obstruction (Class I, Level B)
- residual ASD or VSD with significant left-to-right shunting (Class I, Level B)
- complete AVB (pacemaker implantation) (Class IIa, Level B).
Outcomes and complications
Without surgical repair, only 4% of patients with complete AVSD will survive beyond 5 years of age.63 Conversely, the 10-year survival rate in patients following complete AVSD repair is 83%60 and 93% after partial AVSD repair.64 Table 63.7 summarizes late complications of AVSD, including the most common, left AVV regurgitation (10% of patients).50,60,64
Table 63.7 Late complications of VSDs
Left AVV regurgitation LVOT obstruction Complete AVB Pulmonary vascular disease Atrial or ventricular arrhythmias Left AVV stenosis Right AVV stenosis/regurgitation Residual VSD Aortic regurgitation |
Modified with permission from Craig.50
Left ventricular outflow tract obstruction and bicuspid aortic valve
Anatomic findings and pathophysiology
Left ventricular outflow tract obstructions (LVOTOs) comprise a group of stenotic lesions which may be located at the subvalvar, valvar or supravalvar level. Significant LVOTO creates an increase in afterload, resulting in elevated end-diastolic left ventricular pressure and concentric hypertrophy.
Subvalvar aortic stenosis (SAS)
Fixed SAS ranges from discrete fibrous membrane (91% of cases) to tunnel-like fibromuscular band.65,66 Nearly one-quarter of patients also have bicuspid aortic valve (BAV).65 Other associated cardiac defects include VSD, coarctation of the aorta and Shone syndrome (parachute mitral valve, mitral stenosis, SAS, aortic coarctation, BAV). SAS is usually progressive; AR is common but usually mild.67
Congenital valvar AS
Abnormal valve and cusp formation may result in unicuspid, bicuspid, trileaflet bicommissural or rarely quadricuspid valves. Fifty-four percent of patients with significant AS requiring surgery have congenitally malformed valves, of which more than 90% are bicuspid.68 The estimated incidence of BAV in the general population is 1–2%, affecting more males than females.69 The high incidence (37%) of BAV in first-degree relatives suggests an autosomal dominant inheritance with reduced penetrance.70 BAV is associated with abnormal aortic root tissue structure, including medial disease,71 explaining why patients may develop aortic root dilation and aortic dissection.
Supravalvar AS (SVAS)
SVAS is the less common LVOTO. The morphologic feature of this condition is aortic narrowing at the level of the sinotubular junction. The entire ascending aorta may be involved and, less commonly, the aortic arch and peripheral arteries.66 SVAS is frequently associated with Williams syndrome, which is characterized by intellectual impairment, elfin facies, short stature and, often, peripheral pulmonary artery stenosis. A mutation of elastin gene on chromosome 7q11.23 has been detected in most patients,72 irrespective of the form of SVAS (autosomal dominant form, sporadic form or associated with Williams syndrome).
Clinical presentation
Symptoms may develop in childhood or adulthood, depending on the degree of obstruction. Patients with signon-invasive measurements, or in anificant LVOTO (subvalvar, valvar or supravalvar) may present with dyspnea, angina, syncope, congestive heart failure or signs of endocarditis. The following signs may be found on examination.
- Slow-rising, diminished carotid pulse
- Apex-to-carotid delay (severe)
- Sustained apical impulse
- Ejection click (with BAV, if valve pliable, or with dilated aortic root)
- Diminished aortic component of S2, paradoxic splitting if severe
- S4, S3 if ventricular dysfunction
- Crescendo-decrescendo systolic murmur, late peaking if severe
- Diastolic murmur if AR present
Diagnostic evaluation
Electrocardiogram
Left axis deviation, left ventricular hypertrophy, T-wave inversion with ST segment depression in lateral leads and left atrial enlargement.
Chest radiography
Cardiomegaly (mild, unless significant AR and/or left ventricular dysfunction are present), dilation of ascending aorta (common with BAV), calcifications of the aortic valve.
Echocardiography
Modality of choice to identify and quantify the severity and level of LVOTO (Class I, Level B). Peak instantaneous Doppler velocity and gradient help to determine prognosis and guide the timing of intervention.73,74 Aortic root diameter, left ventricular size/hypertrophy, and systolic/diastolic function should also be assessed.
Exercise stress testing
May be useful if the patient anticipates athletic participation or pregnancy, if clinical findings differ from noninvasive measurements, or in asymptomatic young adults with a mean Doppler gradient greater than 40 mmHg or a peak Doppler gradient greater than 64 mmHg (Class IIa, Level C).
CMR and CT angiography
Both helpful for visualizing the entire intrathoracic aorta and detect associated lesions (Class IIa, Level C).
Cardiac catheterization and angiography
Cardiac catheterization is useful before aortic valve replacement (AVR):75
- when non-invasive imaging is suboptimal or discordant with the clinical evaluation (Class I, Level C)
- to determine if there is co-existent coronary artery disease (CAD) (Class I, Level B)
- to identify the origin of coronary arteries when a Ross procedure (see below) is contemplated, if non-invasive imaging is inadequate (Class I, Level C)
Management
SAS
Surgical resection is the treatment of choice, but the timing of intervention remains controversial. Some authors advocate anearly aggressive approach to prevent AR,65,76,77 while others consider the significant risk of recurrence and are more conservative. Table 63.8 proposes some indications for operative resection.66,74
Table 63.8 Indications for intervention in patients with LVOTO
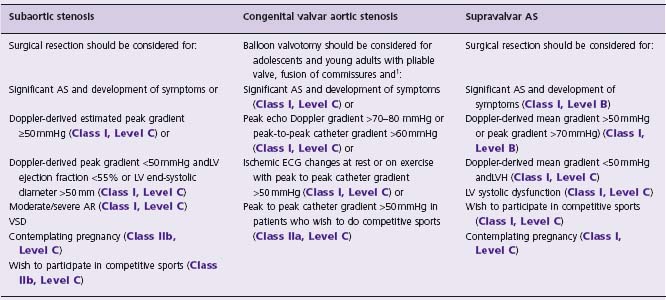
1 Indications for surgical aortic valve replacement in adults with valvar AS are discussed in Chapter 55.
LVH, left ventricular hypertrophy.
Congenital valvar AS
There are currently no proven medical treatments to delay the disease process in patients with AS. It is reasonable to use beta-blockers for dilation of the ascending aorta.66 Management and recommendations for AVR in adults with valvar AS are discussed in Chapter 55.75 Table 63.8 summarizes the indications for balloon valvotomy in adolescents and young adults with congenital valvar AS.75 When valve replacement is required, the Ross procedure may be considered (particularly for young women of reproductive age).
Outcomes and complications
SAS
Recurrence rates following surgical resection vary and are probably close to 15%.65,78,79 Risk factors for recurrence include proximity of the obstructive lesion to the aortic valve and severe obstruction.78 Infective endocarditis and AR are other complications of SAS. AR may develop despite surgical resection of the SAS.66
Congenital valvar AS
It has been suggested that the 3 year-mortality for symptomatic patients with AS, not undergoing surgery, is approximately 75%.80 In asymptomatic patients, however, the risk of sudden cardiac death is less than 1%/year, even in hemodynamically significant AS.73 Complications for patients with LVOTO are listed in Table 63.9.
Table 63.9 Complications of LVOTO (SAS, BAV, SVAS)
Endocarditis Aortic regurgitation Recurrent AS after surgical or balloon intervention in BAV, or following surgical resection in SAS Aortic root dilation or dissection (BAV) Sudden cardiac death Left ventricular failure |
Anatomic findings and pathophysiology
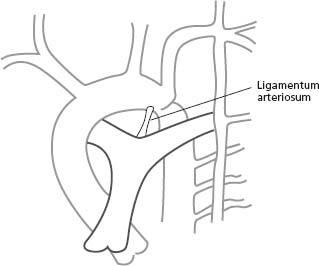
Coarctation of the aorta is a localized narrowing usually located just below the origin of the left subclavian artery (LSCA), in the region of the ligamentum arteriosum (Fig. 63.4). Less commonly, a diffuse form with tubular hypoplasia may involve the aortic arch or the aorta distal to the LSCA. Aortic medial abnormalities may be present.71 Extensive arterial collaterals often develop through the intercostal, subclavian, internal thoracic and scapular arteries to supply the lower body. Aortic coarctation is more frequent in men than women (1.5: 1).66 The reported incidence of BAV in patients with coarctation varies from 27% to 85%.81–83 Coarctation may also occur in conjunction with VSD, mitral valve abnormalities, intracranial aneurysms (in up to 10% of patients84) and Turner syndrome
Clinical presentation
Adults are most often asymptomatic and diagnosis is suspected after fortuitous systemic arterial hypertension or murmurs are found. Other possible clinical presentations include headache, epistaxis, intracranial hemorrhage, leg fatigue on exertion, CHF, angina and aortic dissection.85 Physical examination classically reveals:
- upper body systemic hypertension (blood pressure should be taken in the right arm)
- blood pressure drop between upper and lower extremities
- delayed and weak femoral pulses
- palpable arteries over the scapulae or lateral chest wall
- systolic thrill in suprasternal notch
- sustained apex (pressure overload)
- loud aortic component of S2
- ejection click if BAV ± ejection systolic murmur
- continuous murmurs over the back (collaterals).
Diagnostic evaluation
Electrocardiogram
The ECG may show left atrial and ventricular hypertrophy.
Chest radiography
The so-called “3” sign (double contour in the region of descending aorta) and rib notching (erosion of the posteroinferior border of third to ninth ribs, caused by intercostal collaterals) are two characteristic features on the chest X-ray. The ascending aorta may appear dilated, and overall heart size is normal in the majority of patients.85
Echocardiography
Continuous wave Doppler assessment of the distal aortic arch via the suprasternal notch provides peak pressure gradient through the coarctation. Velocities proximal and distal to the stenosis should be included in an expanded Bernouilli equation to avoid overestimation of the gradient.86 Doppler systolic velocities may be affected by lesion length,87 presence of collateral flow,86,88 and aortic compliance.89 Diastolic velocities should therefore also be measured; the presence of a diastolic tail (elevated diastolic velocities) correlates with significant aortic coarctation.90-92 TTE can also show LVH, associated BAV or dilated ascending aorta.
CMR and CT angiography
CMR and CT angiography are the imaging modalities of choice to evaluate coarctation before and after surgical or transcatheter intervention (site, extent and severity93 of stenosis, presence of aneurysms,94 anatomy of intracranial vessels, and extent of collateral network).
Cardiac catheterization and angiography
A peak-to-peak catheter gradient >20 mmHg is generally accepted as significant coarctation.66 Cardiac catheterization is mainly employed for catheter intervention and for screening the coronary arteries.
Management
Timing of intervention or reintervention (surgery or angioplasty ± stent) in adults with coarctation may be challenging. Nevertheless, intervention should be considered when:
- peak-to-peak catheter gradient is greater than or equal to 20 mmHg across the coarctation (Class I, Level C)
- peak-to-peak catheter gradient is less than 20mmHg across the coarctation in the presence of significant coarctation demonstrated by imaging studies, with radiologic evidence of significant collateral flow.7a,95
The presence of symptoms, systemic hypertension (at rest or with 24-hour monitoring) or LVH should be considered in the intervention decision-making process.
Surgical repair may involve end-to-end anastomosis, interposition graft, patch aortoplasty (abandoned because of the high incidence of aneurysms), ascending to descending aorta conduit repair (for complex/hypoplastic aortic arch) and subclavian flap repair (for neonates). Balloon angioplasty represents an alternative with encouraging results, although more aneurysm formation is reported compared with surgery96 The incidence of aneurysm seems to decrease substantially with primary stent implantation, however.97 Catheter interventions appear to be safe and effective, and thus should be considered for simple native and recurrent coarctation (with catheter gradient >20mmHg) (Class I, Level B).
Table 63.10 Residua following repair of coarctation
Persistent systemic hypertension Recoarctation Aneurysm of the aorta Coronary artery disease AS/AR (if BAV present) Endocarditis/endarteritis Rupture of aortic or cerebral aneurysm Postsurgical repair paraplegia (rare) |
Outcomes and complications
Seventy-five percent of unoperated patients will have died by 46 years of age.66,98 Estimated 30-year survival following repair is 72%.99
Anatomic findings and pathophysiology
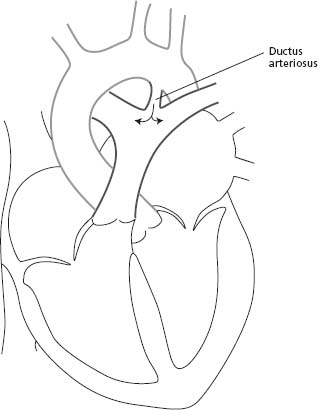
The patent ductus arteriosus (PDA) is a blood vessel that connects the descending aorta (just distal to the LSCA) to the proximal left pulmonary artery (Fig. 63.5). In fetal life, this structure is vital as it carries the blood from the right ventricle to the descending aorta, bypassing the lungs. The ductus normally closes soon after birth. When it remains patent, it can lead to left-to-right shunting with increased pulmonary flow and left heart volume overload.
Clinical presentation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


