Lung Failure |
This chapter focuses on the pathogenesis of acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS). Chapter 141 discusses clinical features and management of these disorders.
A 1994 consensus conference defined the spectrum of ALI/ARDS as follows: (1) ALI, defined as arterial hypoxemia with a PaO2/FIO2 <300, and ARDS, defined as a PaO2/FIO2 <200, accompanied by (2) bilateral pulmonary infiltrates, and (3) absence of left atrial hypertension.1 A more recent report, based on the so-called, “Berlin definition,” recommends identification of three categories of ARDS, based upon the degree of hypoxemia alone: (1) mild ARDS (200 <PaO2/FIO2 ≤300 mm Hg); (2) moderate ARDS (100 <PaO2/FIO2 ≤200 mm Hg); or (3) severe ARDS (PaO2/FIO2 ≤100 mm Hg).2 For the purposes of this chapter, both ALI and ARDS will be referred to as ARDS.
PATHOPHYSIOLOGY OF PULMONARY EDEMA IN ACUTE RESPIRATORY DISTRESS SYNDROME
Pulmonary edema occurs when fluid is filtered into the lungs faster than it can be removed. Accumulation of fluid may have major consequences on lung function because efficient gas exchange cannot occur in fluid-filled alveoli. Lung structure relevant to edema formation and the forces governing fluid and protein movement in the lungs have been the subject of classic and more recent reviews.3
 VASCULAR FLUID AND PROTEIN EXCHANGE
VASCULAR FLUID AND PROTEIN EXCHANGE
The essential factors that govern fluid exchange in the lungs are expressed in the Starling equation for the microvascular barrier:
Jv = LpS[(Pc – Pi) – σd(πc – πi)]
where

The Starling equation predicts the development of two different kinds of pulmonary edema. Increased pressure pulmonary edema occurs when the balance of the driving forces increases, forcing fluid across the barrier at a rate that can no longer be accommodated by lymphatic drainage. Increased permeability pulmonary edema occurs in the presence of ARDS that damages the normal barriers to fluid filtration and allows increased flux of liquid and protein into the extravascular compartments of the lungs. Congestion, atelectasis, and pulmonary edema were features of the original description of the syndrome.4
Thus, pulmonary edema results from increases in either hydrostatic driving pressures (increased pressure edema) or barrier conductance (increased permeability edema), or both. What distinguishes the two types is barrier permeability, which is normal in increased pressure edema, but abnormal in increased permeability edema. Fluid flow into the lungs is driven across the barrier in both types of edema by the balance of pressures. ARDS results primarily from an increase in lung vascular permeability, although some cases may be made worse by the presence of elevated lung vascular hydrostatic pressures.
 INCREASED PERMEABILITY PULMONARY EDEMA
INCREASED PERMEABILITY PULMONARY EDEMA
Increased permeability pulmonary edema is caused by an increase in liquid and protein conductance across the barriers in the lungs. The essential feature is that the integrity of the barrier to fluid and protein flow into the lung interstitium and alveoli is altered. Increased permeability edema is sometimes called noncardiogenic pulmonary edema, and the resulting clinical syndromes in humans are commonly lumped together as ALI or ARDS.
Accumulation of fluid and protein increases when the lung endothelial and epithelial barriers are injured. If the rate of fluid accumulation exceeds the rate at which it can be removed, increased permeability edema occurs. Large animal models employing measurements of hemodynamics and lung lymph flow demonstrate that clinically relevant causes of ARDS, including live bacteria, endotoxin, and microemboli, induce an increase in lung vascular permeability that causes protein-rich lung edema.5–7 Because the barriers limiting fluid and protein flow into the lungs do not function normally when the lungs are injured, the lungs are not protected against edema by the usual safety factors. Although increases in fluid and protein filtration across the lung endothelium can be removed by lymphatics and drained away from the alveolar walls as in increased pressure edema, much more fluid and protein are filtered at any given sum of driving pressures, since the barriers to flow are much less restrictive than normal.
Edema formation in injured lungs is sensitive to hydrostatic driving pressures. Driving pressures are often increased when the lungs are injured because of the vasoconstrictive effects of inflammatory mediators, such as thromboxanes. Thromboxanes may shift the main site of vascular resistance to postcapillary venules, thus increasing hydrostatic pressure at the microvascular fluid exchange sites, or may exert cardiac effects. For example, elevated left atrial pressure, pulmonary venoconstriction, or an increase in cardiac output in sepsis may increase hydrostatic pressure at the microvascular fluid exchange sites. Although a primary event in ARDS is an increase in lung vascular permeability, the magnitude of lung edema formation in ARDS may be substantially increased when lung vascular pressures and volume are elevated, consistent with the effects of elevated hydrostatic pressure on transvascular flux of fluid and protein.
Because the lung endothelial and epithelial barriers are injured in ARDS, the protective effects of protein osmotic pressure differences across them are lost; driving pressure is unopposed by protein osmotic pressure, and even normal hydrostatic pressure results in significant fluid and protein extravasation into the interstitial and alveolar spaces. The ability of the lymphatics to pump the excess filtrate away is increased when the lungs are injured. If the alveolar epithelial barrier is damaged, edema may accumulate readily in alveoli, since most of the resistance to fluid and protein flow into the alveoli is in the epithelial barrier.
To summarize, the majority of patients with ARDS have normal or low hydrostatic pressures and increased alveolar-capillary permeability. However, up to 30% of patients with ARDS may have an elevated left atrial pressure.8 Lung fluid balance is a dynamic concept that incorporates both formation and removal of edema fluid in the interstitium and airspaces.
 LUNG PHYSIOLOGY
LUNG PHYSIOLOGY
The effects of increased permeability edema on lung mechanics and gas exchange depend, in part, on the magnitude of edema accumulation. As with increased pressure edema, the major effects on pulmonary mechanics occur with alveolar flooding.
About 20% to 30% of the extravascular water of the lungs is in the extravascular interstitial tissue. This volume can more than double before alveolar flooding occurs. In experimental lung injury, functional residual capacity (FRC) is decreased as a consequence of alveolar flooding; the loss of units that can be ventilated accounts for most of the decrease in static lung compliance.9 Computed tomography has provided new insights into structure–function relationships in human ARDS. In the early stage of lung injury, when alveolar edema predominates, the lungs are characterized by a more homogeneous alteration of vascular permeability, and edema may accumulate evenly in all lung regions, with a nongravitational distribution. Later in the exudative phase of ARDS, the consolidation is more gravity-dependent. In the organizing phase, lung reticulation appears.
Measurements of pulmonary mechanics in mechanically ventilated patients with ARDS show a decrease in static lung compliance as a consequence of loss of ventilated lung units. In addition, airflow resistance is increased as a result of decreased lung volume.10 Bronchospasm may add to the increase in airflow resistance and may be partially reversed in some patients by administration of inhaled bronchodilators. Chest wall compliance is reduced, probably because of alterations in the intrinsic mechanical properties of the chest wall by abdominal distention, chest wall edema, and pleural effusion. Respiratory mechanics and responses to positive end-expiratory pressure (PEEP) during mechanical ventilation in patients with ARDS originating from pulmonary disease (e.g., pneumonia and associated lung consolidation) and that arising from extrapulmonary disease may differ.11
The injured lung in ARDS may release biologically active substances that can interfere with the normal state of low surface tension in the alveoli. In addition, activated neutrophils may reduce surfactant function in vitro and degrade major surfactant apoproteins through a combination of proteolysis and oxidant radical–mediated mechanisms. Human lung surfactant obtained from bronchoalveolar lavage (BAL) fluid in patients at risk for ARDS and from those with established ARDS are abnormal in chemical composition and functional activity.12 Abnormalities may also be caused by interactions between surfactant and edema proteins, since plasma proteins interfere with surfactant function.
Gas exchange is severely compromised in increased permeability edema, because of both intrapulmonary shunting of blood and ventilation–perfusion inequalities. Clinical evidence indicates that patients with early ARDS have a marked increase in pulmonary dead space fraction.13 This finding indicates that many ventilated lung units are not well perfused, although intrapulmonary shunting may also contribute to the elevated dead space. Minute ventilation is typically twice normal (approximately 12 L/min) at the onset of ARDS.
 PATHOLOGIC FINDINGS
PATHOLOGIC FINDINGS
Based on several studies that included a preponderance of postmortem pathology, the light and electron microscopic appearances of human and animal lung tissue in ARDS have been described.14 The earliest changes are marked by widespread alveolar and interstitial edema, inflammation, and hemorrhage. Hyaline membranes, composed of precipitated plasma proteins, fibrin, and necrotic debris, are frequently found and constitute the footprint of a pathologic finding termed diffuse alveolar damage (DAD), which pathologists use to define ARDS microscopically (Fig. 140-1).15
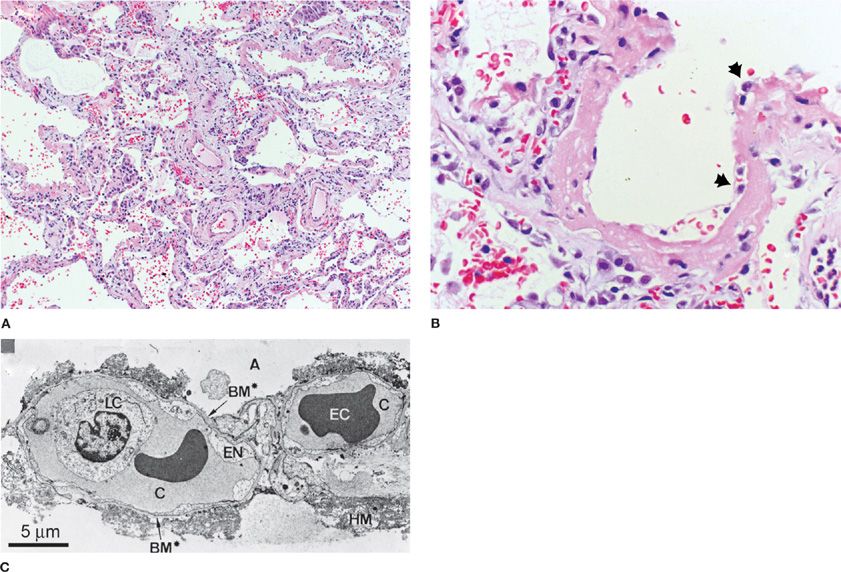
Figure 140-1 A. A low-power light micrograph of lung biopsy specimen collected 2 days after the onset of ALI/ARDS secondary to gram-negative sepsis demonstrates key features of diffuse alveolar damage, including hyaline membranes, inflammation, intra-alveolar red blood cells and neutrophils, and thickening of the alveolar-capillary membrane. B. High-power view of a different field illustrates dense hyaline membrane and diffuse alveolar inflammation. Polymorphonuclear leukocytes are imbedded in the proteinaceous hyaline membrane structure (black arrows). (Histological sections in A and B used with permission of Dr. K. Jones, University of California, San Francisco). C. Electron micrograph from a classic analysis of ALI/ARDS showing injury to capillary endothelium and alveolar epithelium. LC, leukocyte within the capillary lumen; EC, erythrocytes; EN, blebbing of the capillary endothelium; BM, exposed basement membrane where the epithelium has been denuded; C, capillary; A, alveolar space.(C. Reproduced with permission from Bachofen M, Weibel EAR. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982;3(1):35–56.)
The alveolar epithelium may be more extensively damaged than is the vascular endothelium. Alveoli that demonstrate widespread destruction of type I alveolar epithelial cells may appear adjacent to normal alveoli. The injured alveolar epithelium is swollen, disorganized, and, frequently, detached from basement membranes. The alveolar surface may be covered by hyaline membranes. Type I cells are more severely damaged than type II cells. The thin cytoplasmic extensions of cells far from the nucleus, which cover the thin side of the alveolar-capillary barrier, may be most severely affected. The interstitium is widened by edema (especially in peribronchovascular cuffs) and may be filled with leukocytes, platelets, red blood cells, fibrin, and debris (especially near the alveolar walls). The microvascular endothelium may show cytoplasmic swelling or vacuoles and greater numbers of intraluminal leukocytes.
After about 5 to 10 days, the exudative phase of ARDS is followed by a proliferative phase. The relative contributions of the original insult, repair processes, and effects of therapies on this and subsequent phases are not well known. Some abnormalities occurring after the initial exudative phase are related to effects of traditional modes of mechanical ventilation that used tidal volumes between 12 and 15 mL/kg predicted body weight.
Reabsorption of some of the edema fluid characterizes the proliferative phase. Fibrin may be prominent in alveoli and interstitium, and infiltration with inflammatory cells and fibroblasts, which may have been activated very early in the course of lung injury, may be seen. The alveolar epithelium is often cuboidal and made up largely of proliferating type II cells. The air–blood barrier may be thickened by interstitial and epithelial enlargement. The pulmonary vascular bed may be partially or completely disrupted, and structural alterations may reduce its surface area.
In a study addressing the clinicopathologic correlation of the new Berlin definition of ARDS severity with historical autopsy data, a greater proportion of patients with severe ARDS demonstrated histopathologic findings of DAD compared with those with mild or moderate disease.16
 EXPERIMENTAL STUDIES
EXPERIMENTAL STUDIES
The most common clinical disorders associated with the development of ARDS are pneumonia, sepsis, gastric aspiration, and major trauma. Other, less common causes include transfusion-associated lung injury, drug overdose, severe acute pancreatitis, and near drowning. The initiating insult to the lungs occurs either via the airways or the bloodstream.
The exact mechanisms by which the lungs are injured have been studied in humans, animals, and cellular systems. Human studies have provided descriptive data regarding the events that occur in the airspaces before and after the onset of lung injury. Studies using BAL or collection of pulmonary edema fluid in patients following the onset of ARDS have demonstrated a major acute inflammatory response, which begins prior to clinical recognition of ARDS.17 The response peaks during the first 1 to 3 days of clinically defined ARDS and resolves slowly over 7 to 14 days in patients who remain intubated. These studies have shown the complexity of the evolving inflammatory responses, characterized by accumulation of acute response cytokines and their naturally occurring inhibitors, oxidants, proteinases and antiproteinases, lipid mediators, growth factors, and the collagen precursors involved in the repair process.
Hypotheses regarding the mechanisms of lung injury have been tested in animal models and in vitro studies, and several reviews have summarized the findings.3,7 The existing animal models do not completely reproduce all of the aspects of ARDS in humans, in part because human ARDS evolves over a longer period of time than can be studied in the laboratory. In addition, the lungs of humans are exposed not only to the initial injurious insult, but also to the therapies that are used to treat ARDS, such as mechanical ventilation, fluid resuscitation, and antibiotics. Experiments using isolated cells have been helpful in testing specific concepts, but the complexity and redundancy of intact biologic systems is not reproduced in simplified experimental systems. By design, most experimental work limits study to one causative agent, thereby reducing actual clinical complexity to the simplicity of a single experimental pathway. Increased permeability edema in humans is likely to be caused by interactions among a number of different pathways acting in parallel or series, and modulated by process of care variables such as mechanical ventilation, fluid management, vasopressor use, and several comorbidities, including diabetes mellitus, alcohol and cigarette exposure, and renal and liver disease.7
Studies in isolated organs and small animals in which hemodynamic variables are not measured can be difficult to evaluate. Indices of lung injury, usually measured by the appearance of markers in lungs, lavage fluid, or perfusate, are not determined solely by the barrier function of the microvasculature. Indeed, when vascular endothelium is injured, fluid and protein movement from the vascular space into the lungs is sensitive to hydrostatic driving pressures and filtration surface area. Hence, the effects of experimental interventions may be caused by changes in these parameters and not by changes in microvascular barrier function.
The effects of microvascular driving pressures and surface area can be difficult to evaluate, even in large, instrumented animals. In sheep and goats, interpretation of lung lymph fluid and protein flow changes are further complicated by contributions of extrapulmonary lymphatics, physical forces acting on lymphatics, and possible intranodal modification of lymph. Data from experimental animal models suggest that at least two broad categories of mechanisms of ARDS are operative: (1) those that are indirect (i.e., require the participation of intermediary mechanisms, e.g., host defenses); and (2) those that are direct (i.e., do not require intermediary mechanisms; injury probably occurs as a result of contact between an offending substance and lung tissue). These categories overlap, since once the lungs are injured, inflammatory responses occur, which may compound the primary mechanism of injury.
MECHANISMS OF ACUTE RESPIRATORY DISTRESS SYNDROME
The following themes regarding the mechanisms of ARDS are discussed below: infection, inflammation, and direct toxicity. Although discussed separately, they are all interrelated.
 ROLE OF INFECTION
ROLE OF INFECTION
ARDS develops in 20% to 45% of patients with severe sepsis. Increased microvascular permeability to albumin has been shown to accompany human sepsis, and infection and the sepsis syndrome are major causes of ARDS in humans. Patients who develop shock in response to known or suspected infection have a particularly high incidence of ARDS, and the mortality of patients with ARDS associated with infection (i.e., sepsis syndrome) is increased. ARDS also appears to predispose the lungs to infection, and delayed infection is an important cause of morbidity in patients who survive the initial lung insult.
The mechanisms by which infection and sepsis syndrome injure the lungs are only partially understood. The lung injury is likely related to factors other than direct damage by bacteria or other microorganisms, since the prognosis appears unrelated to documented bacteremia or pneumonia. In experimental animals, intravenous infusions of live Pseudomonas aeruginosa, or endotoxins from Escherichia coli, or surgically induced peritonitis result in increased permeability pulmonary edema.18 ARDS caused by endotoxin in sheep is thought to be an inflammatory response mediated, at least in part, by neutrophils and tumor necrosis factor (TNF). Endotoxin may also affect the clotting system and metabolic functions of the lungs, as well as predispose the lungs to the development of pulmonary infections by increasing adherence of bacteria to injured endothelium. Exoproducts of bacteria, such as elastase and Pseudomonas exoenzyme U, also have been shown to injure the lungs.7
Bacterial products may also play a role in pathogenesis of ARDS by sensitizing the lungs to the effects of mechanical stretch. Gram-negative lipopolysaccharide causes an acute inflammatory response in the lungs of humans. Bacterial endotoxin enhances the responses of human alveolar macrophages to positive pressure ventilation; pretreatment of rats with intravenous endotoxin enhances cytokine production in the lungs during mechanical ventilation ex vivo. Furthermore, mechanical ventilation using moderate or large tidal volumes increases the sensitivity of lung macrophages to endotoxin in vitro and the expression of the endotoxin recognition molecule, CD14, on lung cells in vivo. Endotoxin recognition pathways are increased in the lungs of patients with ARDS, and the biologic effects of endotoxin are amplified in the lungs of patients with lung injury. The synergism between bacterial products and mechanical stretch suggests that interrupting these pathways might limit some forms of ARDS in humans.19
Increased permeability edema is associated with impaired antibacterial defenses. In animal models, bacterial infections worsen ARDS. The cause of impaired bacterial defenses in ARDS is not known. Genetic polymorphisms that predispose individuals to the injurious effects of specific bacteria or viruses may influence the development of ARDS. Several polymorphisms are associated with more severe pneumococcal, Legionella, and viral lung infections.20 The genetic factors that regulate the virulence of infecting pathogens also require more research to relate the severity of clinical lung injury to specific microbiologic variables that contribute to severe pneumonia and ARDS.21
 ROLE OF INFLAMMATION
ROLE OF INFLAMMATION
Substantial evidence implicates host defenses and inflammatory responses in the underlying mechanism of ARDS. The balance between protective and injurious innate and adaptive immune responses and hemostatic pathways may determine whether alveolar injury can be repaired and resolved. For example, in lung infection, acute inflammatory responses to pathogens and their toxins cause ARDS through leukocyte protease release, generation of reactive oxygen species, synthesis of chemokines and cytokines, Toll-like receptor engagement, and actions of lipid mediators. However, these same inflammatory mechanisms, when controlled, rather than excessive, are requisite in pathogen containment and clearance.22–24
Normally, the pulmonary circulation contains a large pool of marginated neutrophils that change shape in order to squeeze through the lung capillaries. When neutrophils are activated, they stiffen and become less distensible. These neutrophils are retained for longer periods of time in the pulmonary microcirculation. Endothelial activation leads to increased expression of leukocyte adhesion molecules, providing a second mechanism to slow the transit of neutrophils. Trapped neutrophils respond to chemotactic gradients generated by chemokines produced by alveolar macrophages and mesenchymal cells and migrate into the airspaces. Activated neutrophils generate and release toxic substances (e.g., oxygen metabolites and granular constituents, such as proteases, and cationic lysosomal enzymes) that disrupt the function of the microvascular and epithelial barriers. Normally, these barriers limit liquid and protein flow out of the vascular space and into the alveolar spaces, mitigating the development of permeability edema.19
Inflammatory responses also have the potential to induce lung cell injury by activating cell death pathways, leading to apoptosis. Bacterial products, such as Pseudomonas Exoenzyme U and mechanical stretch, may lead to direct cellular necrosis. Apoptosis is mediated by a family of death receptors, including TNF and Fas receptors. The Fas ligand (FasL) is a 45-kD peptide that is shed from the cell surface by the action of metalloproteinases.25 Biologically active soluble FasL (sFasL) accumulates in the lungs of patients with ARDS, inducing apoptotic death of human lung epithelial cells in vitro. Human sFasL induces epithelial cell death in the lungs of rabbits; a monoclonal antibody that activates membrane Fas causes alveolar wall apoptosis and fibrosis in the lungs of mice.25
Apoptosis and inflammation pathways intersect, as stimulation of membrane Fas induces cytokine production in human macrophages and inflammation in the lungs of rabbits and mice. In addition, lung injury may be able to trigger apoptosis pathways in distant organs, such as the kidney, perhaps by increasing the concentrations of circulating sFasL. Thus, inflammatory responses may trigger cell death pathways, and cell death pathways triggered by sFasL may induce inflammation in the lung alveolar environment. Recent human studies implicate apoptosis as an important mechanism in human lung injury.26
 ROLE OF DIRECT TOXICITY
ROLE OF DIRECT TOXICITY
Inflammation is not required for all forms of ARDS. ARDS may develop in neutropenic patients. A clinical trial using granulocyte colony–stimulating factor to increase the number and activation state of circulating neutrophils in patients with severe pneumonia did not reveal an increased incidence of ARDS.27 Lung injuries that do not require the participation of neutrophils have been described in animal models.
Direct lung injury is also thought to occur in humans. Putative agents that directly injure the lungs include mechanical forces during mechanical ventilation, toxic and corrosive chemicals and gases (e.g., hydrochloric acid, chlorine gas, phosgene), and aspiration of fresh or salt water (near-drowning). Many of these injuries develop rapidly, supporting the idea that injury is caused directly by contact with the respiratory epithelium in the airways or alveolar walls.
Inflammatory pathways are likely to be rapidly activated following many types of direct lung injury, as probably occurs following aspiration of gastric secretions – one of the most common clinical causes of ARDS. Lung injury occurs rapidly, especially to the epithelium. The injury is probably related, in part, to the low pH of the aspirated stomach contents. Aspirated acid is almost immediately neutralized. However, within hours, proinflammatory mediators are released, the injured lung is infiltrated with neutrophils, fibrin accumulates in the alveolar spaces, and further structural damage is seen on histologic examination. Thus, even direct toxic injuries damage the lung in part through activation of inflammatory pathways.
 BIOLOGIC MARKERS
BIOLOGIC MARKERS
Considerable interest exists in finding a simple test of blood, urine, or BAL fluid that would identify patients at risk for, or in the earliest stages of, ARDS, or that might predict clinical outcome.
Although products of complement activation have been proposed as markers, their serum levels correlate poorly with lung injury. Measurement of circulating endotoxin is not appropriately sensitive or specific for the presence or risk of developing lung injury. The same is true for measurements of release or activity of angiotensin-converting enzyme.28 Von Willebrand factor (VWF) antigen may be useful as a plasma marker of impending ARDS in patients with nonpulmonary sepsis.29 VWF levels are elevated in the edema fluid and plasma of patients with ARDS and correlate with poor clinical outcomes.30 Also, new work indicates that elevated levels of plasma angiopoietin-2 predict the development of ALI in patients with sepsis identified in the emergency department.31 While increases in other biochemical and inflammatory markers, including surfactant protein D and interleukin-6, correlate somewhat with lung injury and mortality, no simple biologic marker currently serves in the same diagnostic capacity, as do cardiac enzymes in evaluation of suspected acute myocardial infarction.32
Other mediators of inflammation in ARDS have been studied. For example, increased levels of TNF are detected in blood and BAL fluid in lung injury, but an association between TNF levels and the development of ARDS has not been found. Furthermore, elevated TNF levels are found in patients with severe congestive heart failure. Lipoxygenase products of arachidonic acid metabolism have been detected in pulmonary edema fluid, BAL fluid, plasma, and urine, and elastase has been detected in BAL fluid in the setting of lung injury.
ARDS follows a wide variety of insults of varying severity. In a study of injured patients with ARDS, investigators found increased levels of IL-6 and thrombomodulin in those who had fewer ventilator-free days.33 However, since ARDS is such a heterogeneous disorder, with so many possible mechanisms, it is unlikely that any single marker that unequivocally identifies the risk or the presence of ARDS will be found. An investigative focus on particular subgroups of patients with common causes of lung injury, coupled with study of much larger groups of more precisely diagnosed patients, may be a better pathway for future research.
Figure 140-2 depicts multiple pathways involved in the pathogenesis of ARDS in the context of normal and injured alveoli. The depictions place emphasis on potential pathways for injury across the vascular endothelium and alveolar epithelium.
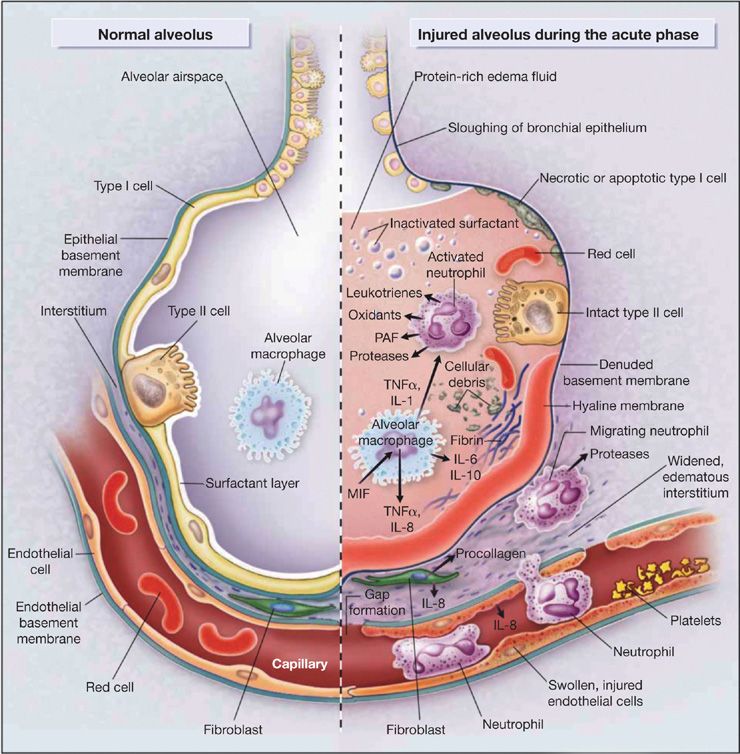
Figure 140-2 Multiple cellular responses and mediators contribute to alveolar-capillary membrane injury (right-hand side) and the transition from normal alveolar structure and function (left-hand side) in the acute phase of ALI/ARDS. Original investigations of the pathogenesis of ALI/ARDS searched for single mediators that provided final common pathways to inflammation and alveolar edema. Current concepts of pathogenesis involve multiple molecular factors of several classes, a variety of responding cells, and an imbalance between injurious and reparative signals and pathways. See text and Ware & Matthay (2000). (Reproduced with permission from Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349.)
VENTILATOR-INDUCED LUNG INJURY
The most important development of the last 10 years in our understanding of the pathogenesis and treatment of ARDS is recognition that the long-standing practice of mechanically ventilating affected patients using high tidal volumes and airway pressures worsens the injury. Animal studies first suggested the potential contributory role of high tidal volumes and elevated airway pressures in the pathogenesis of lung injury; subsequently, clinical trials confirmed the findings.3
 ANIMAL STUDIES
ANIMAL STUDIES
Experimental studies have shown that ventilation using high tidal volumes may increase vascular filtration pressures; produce stress fractures of microvascular endothelium, alveolar epithelium, and basement membranes; and cause lung rupture (so-called ventilation-induced lung injury).34 The injury appears to be due to increased lung excursions at high volumes (volutrauma), rather than the high-airway pressure, per se, since it can be prevented by limiting thoracic motion (e.g., by placing the chest in a cast).
The concept of volutrauma was first established in 1974 when investigators found that modestly elevated tidal volumes, especially in the absence of PEEP, caused lung edema in rats.35 Several years later, additional animal studies further demonstrated the potential injurious role of high tidal volumes and elevated airway pressures, an effect termed ventilator-induced lung injury (VILI).36 Subsequent experiments demonstrated that VILI could also induce release of several proinflammatory cytokines, injuring the lung and other organs – a process referred to as “biotrauma.”37 These animal studies stimulated clinical investigation that revolutionized the care of patients with ARDS.36
 CLINICAL STUDIES
CLINICAL STUDIES
The compelling evidence from animal experiments and small clinical trials prompted clinical studies aimed at testing the potential benefit of lower tidal volumes and reduced airway pressures during mechanical ventilation in management of ARDS. In a large, multicenter, National Heart Lung and Blood–sponsored trial of 861 patients, mortality was reduced from 40% to 31% using a tidal volume of 6 mL/kg/ideal body weight and a limited plateau airway pressure of less than 30 cmH2O.38 In this trial, use of small tidal volumes was associated with a lower incidence of nonpulmonary organ failure. The protocol for carrying out the lung protective ventilatory strategy is described in detail in Table 140-1. The results of the trial transformed the management of patients with ARDS. Notably, a follow-up clinical trial demonstrated that ventilation using the limited tidal volumes and plateau pressure of the original study is associated with an overall reduction of mortality to 26%. In the follow-up trial, although elevated levels of PEEP did not decrease mortality, the basic lung protective strategy was validated as effective.



