Acute Myocardial Infarction: Complications
Judith S. Hochman
Overview
Complications of myocardial infarction (MI) include (a) pump failure [left ventricular [LV] or right ventricular (RV)], which is the leading cause of death in hospitalized MI patients; (b) LV aneurysm; (c) systemic emboli; (d) reinfarction (infarct extension); (e) ischemia; (f) myocardial rupture (free wall, ventricular septal, and papillary muscle); (g) pericardial effusion; and (h) pericarditis. Except for reinfarction and hemorrhagic complications, reperfusion therapy reduces the incidence of most complications.
Echocardiography with color flow Doppler is an excellent tool for rapidly assessing all complications and for evaluating hypotension, congestive heart failure (CHF), and cardiogenic shock. Conditions that mimic shock complicating acute MI, such as aortic dissection, can be readily detected. Management of each complication must be related to its pathophysiology. Mechanical problems, such as ventricular septal rupture (VSR) and mitral regurgitation (MR), should be surgically corrected. Increasing use of early revascularization for cardiogenic shock, based on randomized trial evidence demonstrating improved survival, has resulted in decreasing mortality rates over time in large, community-based studies of patients in cardiogenic shock. Nevertheless, the mortality rate remains high. Early reinfarction, which is associated with increased mortality, may be managed preferentially with percutaneous coronary intervention (PCI) or thrombolysis when ST elevations occur. Future efforts must focus on preventing complications, because mortality rates are high once complications develop.
General Pathophysiology
The spectrum of complications of myocardial necrosis, from CHF to arrhythmias to post-MI angina, is illustrated in Figure 20.1 (1). Complications of MI tend to occur when infarcts are large and extensively transmural, in the absence of tissue perfusion due to microvasculature obstruction (2,3,4). A large, transmural infarct is more prone to expansion (i.e., thinning and dilation) with its attendant increased risk for myocardial
rupture, LV aneurysm, LV thrombus, pump failure, and pericarditis (see Fig. 20.1) (1). Refractory, sustained ventricular tachycardia develops after MI most frequently in relation to large transmural infarcts that result in LV dilation. Although infarct size is a major determinant of complications, certain strategic locations of small, extensively transmural infarcts can result in devastating complications, such as LV free-wall rupture or rupture of the posteromedial papillary muscle, even when expansion does not occur (Fig. 20.1) (1). Indeed, in an autopsy study of patients dying of acute MI, infarct size was substantially smaller in patients with cardiac rupture than in patients dying of primary pump function or arrhythmias. An anteroapical location of MI increases the likelihood of acute infarct expansion and mural thrombosis. Massive acute myocardial necrosis, multiple small infarcts (acute and prior), and recurrent ischemia or reinfarction may each lead to cardiogenic shock (Fig. 30.1) (1). New myocardial necrosis or infarct extension (i.e., reinfarction) may occur after the initial MI and result in a larger total area of infarction. In contrast, infarct expansion involves no additional myocardial necrosis, but results in a larger functional infarct size with a greater percentage of the LV being composed of necrotic myocardium or scar (5). These two complications are compared in Fig. 20.1 (1). Extent and severity of disease in non–infarct-related coronary arteries and important characteristics of the patient—including age, gender, and comorbidities (e.g., diabetes mellitus and prior hypertension)—also contribute significantly to the development of acute MI complications. These complications are not entirely independent, and each can lead to other complications.
rupture, LV aneurysm, LV thrombus, pump failure, and pericarditis (see Fig. 20.1) (1). Refractory, sustained ventricular tachycardia develops after MI most frequently in relation to large transmural infarcts that result in LV dilation. Although infarct size is a major determinant of complications, certain strategic locations of small, extensively transmural infarcts can result in devastating complications, such as LV free-wall rupture or rupture of the posteromedial papillary muscle, even when expansion does not occur (Fig. 20.1) (1). Indeed, in an autopsy study of patients dying of acute MI, infarct size was substantially smaller in patients with cardiac rupture than in patients dying of primary pump function or arrhythmias. An anteroapical location of MI increases the likelihood of acute infarct expansion and mural thrombosis. Massive acute myocardial necrosis, multiple small infarcts (acute and prior), and recurrent ischemia or reinfarction may each lead to cardiogenic shock (Fig. 30.1) (1). New myocardial necrosis or infarct extension (i.e., reinfarction) may occur after the initial MI and result in a larger total area of infarction. In contrast, infarct expansion involves no additional myocardial necrosis, but results in a larger functional infarct size with a greater percentage of the LV being composed of necrotic myocardium or scar (5). These two complications are compared in Fig. 20.1 (1). Extent and severity of disease in non–infarct-related coronary arteries and important characteristics of the patient—including age, gender, and comorbidities (e.g., diabetes mellitus and prior hypertension)—also contribute significantly to the development of acute MI complications. These complications are not entirely independent, and each can lead to other complications.
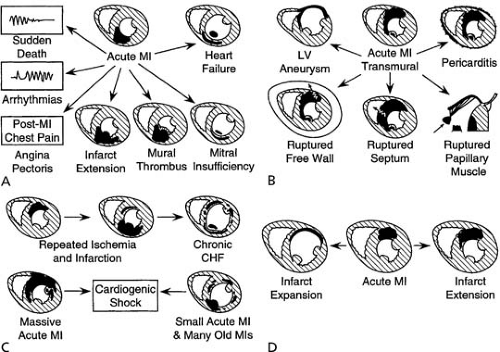 FIGURE 20.1. Complications of MI (schematic). (A) General complications. (B) Complications of transmural infarctions. (C) Cardiogenic shock as a result of either a massive acute infarction or a small acute process in a heart already involved by multiple old infarctions. (D) Comparison of infarct expansion (aneurysm formation) and infarct extension (reinfarction). Abbreviations: CHF, congestive heart failure; LV, left ventricle. (Source: From Edwards WD. Pathology of myocardial infarction and reperfusion. In: Gersh BJ, Rahimtoola SH, eds. Acute myocardial infarction. Boston: Chapman & Hall, 1996:16–50 , with permission.) |
Infarct Expansion, Left Ventricular Remodeling, and Left Ventricular Aneurysm
Pathophysiology
LV remodeling after MI refers to structural and functional changes in the infarct zone and in remote, uninfarcted myocardium that begin minutes after MI and may continue for months or years. These changes include (a) infarct expansion (thinning and dilation of the infarct zone); (b) subsequent dilation of the remote, uninfarcted myocardium with hypertrophy; (c) interstitial fibrosis and impairment of contraction; and (d) global change from a normal LV, elongated ellipse to a more spherical shape (6,7,8,9,10). Neurohormonal mediators and matrix metalloproteinases play an important role in this process (11). LV aneurysm, a discrete bulge of the LV composed of fibrotic tissue, results when severe infarct expansion persists and scar is laid down on the topographic substrate.
Infarct Expansion and Left Ventricular Remodeling
Infarct expansion occurs soon after onset of coronary occlusion (6,12). It is reversible if coronary flow is reestablished rapidly; however, it may progress in a time-dependent manner if flow is not reestablished or is reestablished late (13). Absence of myocardial perfusion at the microvascular level, even in the presence of Thrombolysis in Myocardial Infarction (TIMI) III flow, is associated with progressive LV dilation (2,3). Infarct size is a major determinant, with large infarcts expanding more frequently and more severely than small ones (7,9). Anterior infarcts and those involving the apex are at greatest risk for infarct expansion (5,6), probably as a result of a thinner LV wall and greater radius of curvature at the apex, which increase stress on the LV wall per Laplace’s equation. Transmural infarcts are more likely to expand; nontransmural infarcts are largely protected from expansion (7,13). Infarct expansion is the pathologic substrate for type III cardiac rupture. Disruption of the intercellular collagen matrix may lead to slippage of sheets of myofibrils and expansion of the affected area (10,15).
Infarct expansion results in early LV dilation (6,7) and MR (see Mitral Regurgitation). It is associated with CHF and LV intracavitary thrombosis (8,15,16). Long-term global remodeling with dilation and impairment of uninfarcted myocardial segment contraction begins soon after a large infarction and progresses for months to years (9,10). LV volume strongly correlates with long-term mortality (17,18).
Left Ventricular Aneurysm
Regional expansion of the infarct zone that results in a discrete diastolic and systolic bulge and that is not reversed by
reperfusion or other measures produces chronic, true LV aneurysm. Early LV aneurysms may be referred to as functional aneurysms (20) because they may be reversible. In time, scar tissue is laid down on the already spatially deformed infarct zone. Pathologic examination of LV aneurysm reveals primary scar tissue (19,22); however, LV aneurysms resected years after MI sometimes contain viable and presumably hibernating myocardium (21). In contrast to true LV aneurysm, pseudoaneurysm or false LV aneurysm represents localized rupture of the myocardium and is discussed later in this chapter (22).
reperfusion or other measures produces chronic, true LV aneurysm. Early LV aneurysms may be referred to as functional aneurysms (20) because they may be reversible. In time, scar tissue is laid down on the already spatially deformed infarct zone. Pathologic examination of LV aneurysm reveals primary scar tissue (19,22); however, LV aneurysms resected years after MI sometimes contain viable and presumably hibernating myocardium (21). In contrast to true LV aneurysm, pseudoaneurysm or false LV aneurysm represents localized rupture of the myocardium and is discussed later in this chapter (22).
Left Ventricular Thrombus
Inflammation of the endocardium resulting from myocardial necrosis in any location may produce layered, mural thrombus; however, thrombus most frequently develops in anterior infarcts (16,23) with expansion or aneurysmal dilation that involves the apex, caused by the combination of endocardial inflammation and stasis. More extensive thrombi with a protruding appearance are at increased risk for systemic embolization (16), and they typically occur in the apex.
Clinical Profile
Risk Factors
Patients with myocardial infarcts that involve the apex of the left ventricle, particularly those with anteroapical, Q-wave infarcts are at greatest risk for infarct expansion and aneurysm formation (5,6). A discrete, posterobasal aneurysm may less frequently develop following inferoposterior MI. Patients who do not receive reperfusion therapy or in whom reperfusion therapy fails to reestablish myocardial perfusion are at greater risk for aneurysm formation.
High ventricular afterload states, such as hypertension during acute MI and elevated plasma angiotensin-converting enzyme (ACE) activity levels, are associated with infarct expansion (15,24,25). Administration of corticosteroids after acute MI is associated with delayed infarct healing and development of LV aneurysms (26). Both corticosteroids and nonsteroidal antiinflammatory drugs have been demonstrated experimentally to induce infarct expansion when administered acutely after coronary occlusion (27,28,29).
Diagnosis
Clinical Findings
The classic physical finding of LV aneurysm is a dyskinetic apical impulse on palpation. The electrogradiogram (ECG) typically shows an extensive, anteroapical Q-wave MI with persistent ST-segment elevation. Persistent T-wave inversion is also a marker of progressive LV dilation (30). Chest roentgenography is insensitive but typically demonstrates a bulge in the LV silhouette.
Imaging
On imaging, LV aneurysm is defined as a discrete bulge in the LV contour during both diastole and systole, which typically exhibits dyskinetic (i.e., paradoxical) expansion during systole. Echocardiography with color flow Doppler is the imaging modality of choice and can be used to distinguish discrete LV aneurysms from false aneurysms. Other techniques to assess an aneurysm include biplane left ventriculography, radionuclide ventriculography, computed tomography, and magnetic resonance imaging (MRI).
Clinical Consequences
Cardiac Rupture, Congestive Heart Failure, and Arrhythmias
Rupture of the myocardium may result from acute infarct expansion, but it does not occur in chronic LV aneurysms with scar. Acute CHF complicating MI more frequently occurs when infarct expansion is demonstrated (8,15). Infarct expansion and acute aneurysmal dilation may result in CHF caused by paradoxic systolic bulging with reduced mechanical efficiency of the left ventricle (i.e., wasted work) (21) and elevated wall stress in the remote, uninfarcted myocardium, causing global ischemia. Chronic LV aneurysms are associated with chronic CHF. In this context, the extent of systolic bulging with chronic scar is minimal and wasted work only plays a significant role when the aneurysm is composed of viable myocardium interspersed with scar tissue or of thin scar that expands with each systole (22).
Recurrent and sustained monomorphic ventricular tachycardia may occur in acute infarct expansion or in chronic LV aneurysm and may be refractory to antiarrhythmic therapy (see Chapter 66). LV volume correlates strongly with short- and long-term mortality (17,18), with sudden cardiac death occurring at least as frequently as death from progressive pump failure. Early LV dilation results in late progressive LV enlargement with associated CHF and malignant ventricular arrhythmias late post-MI (31).
Left Ventricular Thrombi, Systemic Emboli, and Stroke
Layered mural and protruding thrombi typically develop within the first week following acute MIs with expanded or aneurysmal akinetic or dyskinetic segments, especially those involving the LV anterior wall and apex. Because LV thrombi more frequently occur with large infarctions they have become less common in the reperfusion era. Recent data suggest that 4% to 8% of all MI patients develop LV thrombus (32,33,34). Risk factors for LV thrombus are Killip class greater than I, ejection fraction (EF) less than 40%, anterior infarction, no reperfusion, and early intravenous β-blocker use (23). LV thrombi are associated with increased risk for systemic embolization, particularly when they have a protruding appearance (16). Stroke is the primary manifestation of cardiac emboli (occurring in 85% of cases); however, the overall incidence of arterial embolism is low (35,36,37,38), whether LV thrombus is visualized or not. The reported incidence (38) of systemic emboli associated with anterior MI in the prereperfusion era was 2% to 6%. Patients with atrial fibrillation (AF) after MI are at increased risk for systemic emboli from left atrial thrombi. Patients with chronic LV aneurysm are at low risk for systemic embolization (39), perhaps because the aneurysmal area containing thrombus is noncontractile and there is no longer endocardial inflammation. However, when global LVEF is reduced after MI (<40%), the rate of stroke is 1.5% per year even when CHF is not present. The risk increases with decreasing EF and older age (40). Patients with LVEF less than 28% have a 5-year stroke rate of 8.9%. However, many of these strokes are likely caused by cerebrovascular ulcerated plaques (as opposed to cardiac emboli) given the evidence that multiple ulcerated plaques occur throughout the vasculature.
Management
Medical Therapy
Inhibition of the Renin/Angiotensin/Aldosterone Axis
ACE inhibitors have been shown in experimental models and in clinical investigations (31,41,42) to reduce acute infarct expansion and progressive LV remodeling. When successful reperfusion has inhibited infarct expansion and aneurysm formation, ACE inhibitors have not been clearly shown to limit expansion further (43). A detailed discussion of the use of ACE inhibitors in MI can be found elsewhere in this textbook (see Chapters 19 and 21). Because infarct expansion begins early, therapy should be instituted within the first 24 hours of onset of MI in patients at risk for expansion and aneurysms (i.e., those with nonreperfused or large anteroapical Q-wave MIs, particularly with associated hypertension). A short-acting ACE inhibitor is recommended for initial therapy (e.g., captopril), so that it can be discontinued if hypotension develops. The initial dose should be 6.25 mg followed by 12.5 mg, then 25 to 50 mg every 8 hours as tolerated. After several days, the patient may be switched to a long-acting ACE inhibitor. Aldosterone antagonists and angiotensin receptor blockers also reduce adverse LV remodeling in experimental models, but have not been extensively clinically tested within 24 hours of acute MI (44,45). The use of these agents in patients with pulmonary congestion is discussed in the section on Acute Left Ventricular Failure.
Anticoagulation: Therapy for Left Ventricular Thrombus
Full oral anticoagulation for 3 months significantly accelerated the resolution of LV thrombus in a small randomized trial (46) of acenocoumarol versus sulfinpyrazone versus placebo. Full-dose warfarin is recommended when LV thrombus is visualized, although there are no data for this subset from randomized trials.
Anticoagulation: Prevention of Systemic Emboli
In the prereperfusion era, when patients were hospitalized for weeks, anticoagulant therapy reduced the occurrence of systemic and pulmonary embolism (PE) (47,48). The 30-day rate of stroke in a largely reperfused ST-elevation MI population was not reduced by heparin (reviparin) compared to placebo in the context of therapy with aspirin (ASA) in 97% and thienopyridines in 50% (49).
The Survival and Ventricular Enlargement investigators (40) reported that long-term use of warfarin in patients with EF less than 40% after MI was strongly associated with a reduced 5-year stroke rate. Of note, ASA was less strongly associated with a reduced risk (40). In the Anticoagulants in the Secondary Prevention of Events in Coronary Thrombosis-2 (51) and Warfarin-Aspirin Reinfarction Study-2 (52) randomized trials, warfarin alone or in combination with ASA was superior to ASA alone post-MI in preventing stroke as well as MI and death (52). However, stroke, MI, and death were not reduced by lower intensity warfarin (Coumadin) plus ASA compared with ASA alone in the Combined Hemotherapy and Mortality Prevention (53), the Low-dose Warfarin and Aspirin Study (54), or the Coumadin Aspirin Reinfarction Study (54,55).
Anticoagulant Regimens
Warfarin dosing
Warfarin therapy (full dosing or moderate intensity when combined with ASA) is recommended following infarction in patients with documented LV thrombus, systemic emboli and in those with AF (56,57). It is recommended that patients with these high-risk characteristics be treated with warfarin indefinitely if they are not at increased risk of bleeding. It is also reasonable to use warfarin for patients with extensive wall motion abnormalities, particularly those with large anterior MI or apical akinesis or dyskinesis, for 3 months or indefinitely in those who are not at increased risk of bleeding. This approach is supported by the Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis 2 and Warfarin Reinfarction Study (WARIS) II studies, which demonstrated reduction of vascular events, stroke, death, and MI with high-intensity warfarin (international normalized ratio [INR] 2.8–4.0) or moderate-intensity warfarin (INR 2.0–2.5) plus ASA compared to ASA alone, albeit at the expense of excess bleeding (51,52). However, lower intensity warfarin plus ASA compared with ASA alone resulted in similar event rates in three trials (53,54,55).
Reinfarction and Ischemia
Pathophysiology
Importance of Coronary Perfusion Pressure
It is critical to maintain coronary perfusion pressure during acute MI to facilitate reperfusion and sustain patency of the Infarct related artery (IRA) postfibrinolysis or post-PCI (58,59). There is evidence of worse outcomes and larger MIs when hypotension occurs with use of nitrates and ACE inhibitors early in acute MI (60,61).
Clinical Profile
Diagnosis
The incidence of reinfarction or infarct extension varies widely, depending on the clinical definition and the diligence with which it is sought. ECG changes alone are the least specific method of diagnosing infarct extension, and they markedly overestimate its occurrence (5). ST-segment elevation that progresses days after MI may represent pericarditis or infarct expansion. Evolution of deepening T waves is typical after MI and does not usually represent ischemia or infarct extension. Recurrent chest pain is also nonspecific, with angina or pericarditis as other causes of post-MI chest pain. Recurrent elevations in creatine kinase with muscle and brain subunits (CK-MB) after its disappearance or to higher than 50% of the prior value are regarded as the standard for reinfarction. Because troponin I and T may remain elevated for up to 2 weeks, they are not used to diagnose reinfarction (56). The full constellation of recurrent chest discomfort and recurrent elevation in ST segments followed by recurrent elevation in CK-MB infrequently occurs but constitutes the typical diagnosis, both in large clinical trials and in clinical practice. We recommend obtaining daily CK-MB measurements for 2 days after MI as well as 8-hour samplings for 24 hours if a recurrent event develops (e.g., chest pain, worsening ST elevations or depressions).
Incidence, Risk Factors, and Prognosis
The incidence of early reinfarction varies depending on the initial therapy and ranges from 16% in observational studies of nonreperfused patients to less than 5% in randomized clinical trials (see Chapter 19).
Prevention
Recurrent MI is, in part, related to the early therapy used in treating the initial MI; this is discussed in Chapter 19.
Management
Reinfarction is managed using the same medications as those used for the initial ST-elevation or non–ST-elevation MI (see Chapters 18 and 19), including ASA, heparin (low-molecular-weight or unfractionated heparin), clopidogrel, GP IIb/IIIa antagonists, and β-blockers, but with the caveat that patients with recurrent ischemia are a high-risk group who merit early cardiac catheterization. Readministration of thrombolytics for recurrent MI with recurrent ST-segment elevations on ECG has been used (65), with high successful response rates. Direct PCI should be performed, if available, for recurrent coronary artery occlusion.
Calcium channel blockers may be added if recurrent angina occurs on maximal doses of β-blockers and nitrates. Intraaortic balloon pump (IABP) counterpulsation should be used for patients with ischemia and hemodynamic instability (i.e., hypotension, hypoperfusion, or low cardiac output) and for refractory ischemia if revascularization facilities are not immediately available.
Reinfarction and postinfarction ischemia identify a high-risk subgroup of patients in whom urgent cardiac catheterization is indicated with prompt revascularization with PCI or coronary artery bypass grafting (CABG) to reduce the risk of long-term events, depending on the coronary anatomy, LV function, and the extent of myocardial jeopardy.
Acute Left Ventricular Failure
Pathophysiologic Principles
General
LV failure in acute MI varies widely in terms of pathophysiology and severity. Mild-to-severe pulmonary congestion may occur alone or in association with depressed stroke volume (SV) and cardiac output (66,67). Diastolic or systolic dysfunction may predominate. At the severe end of the spectrum of pump failure, cardiogenic shock is a state of severe tissue hypoperfusion caused by cardiac dysfunction.
Congestive Heart Failure
After acute coronary occlusion, LV systolic and diastolic function changes over minutes, hours, and weeks. Superimposed fixed or transient MR, recurrent ischemia, or reinfarction may have a major effect. Compliance of the infarct zone evolves over time (68), and stunned myocardium may recover function (69,70). Elevations in LV filling pressures resulting from systolic or diastolic dysfunction may reduce the pressure gradient that maintains coronary blood flow and thereby initiate a cycle of hypoperfusion and worsening LV dysfunction (Fig. 20.2) (71).
Systolic Dysfunction
The composite effect of dysfunctional myocardial segments, as well as the hyperkinetic or normokinetic motion of the uninfarcted myocardium, determines global LV function. These abnormalities may be old (e.g., caused by prior infarctions) or new and may be reversible or irreversible. Global LV function is the strongest determinant of pump failure and death caused by MI (72,73,74). Lack of compensatory hyperkinesis of uninfarcted zones correlates with the development of pump failure. The LV typically dilates when significant systolic dysfunction is present, caused by early infarct expansion and the effect of the resultant increased wall stress on the remote myocardium. LV dilation plays an acute compensatory role by allowing the high end-diastolic volume (EDV) to maintain SV when EF is significantly depressed. This acute adaptive effect is counterbalanced by deleterious long-term effects of LV dilation including increased wall stress, hypertrophy, and further dilation, followed by failure of the remote myocardium.
The clinical manifestations of CHF when LV systolic function is depressed vary. Neurohormonal (75,76,77) (see Fig. 20.2) and hydration status may explain why some patients with low EFs have pulmonary congestion and others do not (78). Diastolic dysfunction owing to infarction contributes substantially to increased filling pressure. Furthermore, changes in peripheral circulation and in end organs, particularly in patients with prior LV damage, may influence the symptomatic response to the acute insult.
Recovery of contractile function in akinetic and even dyskinetic segments following acute MI is well documented and is caused by resolution of myocardial stunning and hibernation. Stunning may take weeks to months to resolve, and function of hibernating myocardium is only restored when coronary flow is normalized. Even late reperfusion may improve wall motion and global EF.
Diastolic Dysfunction
Diastolic dysfunction with reduced LV compliance is a relatively common cause of pulmonary congestion in acute MI. Ischemia causes impaired myocardial relaxation (79). Severe pulmonary edema may result from global subendocardial ischemia caused by three-vessel or left main coronary artery disease (CAD) with a normal heart size. Additionally, the infarct zone undergoes alterations in compliance that result in varying LV distensibility over time following coronary occlusion (68). Reperfusion into areas of irreversible necrosis leads to contraction band necrosis, hemorrhage, and edema, resulting in acutely stiff infarcts zones (80) and, therefore, reduced LV compliance.
Diastolic abnormalities of the LV that impede LV filling may result in both an increase in LV filling pressures and a decrease in SV in the absence of global LV systolic dysfunction. Preexisting conditions that may cause reductions in ventricular compliance, such as hypertension and diabetes, result in an even greater shift of the pressure–volume curve when ischemia or infarction is superimposed.
Cardiogenic Shock Caused by Primary Left Ventricular Failure
LV failure is the most common cause of cardiogenic shock complicating acute MI. The prospective SHOCK Trial Registry identified LV failure as the etiology of shock in 79% of patients (81).
Patients with acute MI who develop cardiogenic shock typically have severe atherosclerotic disease involving all three major coronary arteries and often a severe stenosis in the Left anterior descending (LAD) coronary artery. Significant stenosis in the left main coronary artery is seen in 16% to 21% of patients (82,83,84). Prior MI is present in approximately 40% of patients; when an old infarct is extensive, even a small, new infarction may lead to shock. Although autopsy studies traditionally have demonstrated that at least 40% of the LV mass is affected in patients dying from cardiogenic shock complicating MI (83,85), recent evidence demonstrates that shock can
occur because of reversible ischemia or because of a mismatch between inappropriate systemic vasodilation and reduced cardiac output due to LV dysfunction. Evidence for elevated levels of inflammatory markers and adverse effects of inducible nitric oxide synthase suggest that a systemic inflammatory response syndrome and high levels of nitric oxide seen in other etiologies of shock, play a role in the genesis and persistence of cardiogenic shock (87,88,89,90,91) (see Fig. 20.2). There is a wide range of EFs demonstrated on patients with cardiogenic shock due to LV failure (average LVEF ∼30%, range <10% to >50%) (92,93,94).
occur because of reversible ischemia or because of a mismatch between inappropriate systemic vasodilation and reduced cardiac output due to LV dysfunction. Evidence for elevated levels of inflammatory markers and adverse effects of inducible nitric oxide synthase suggest that a systemic inflammatory response syndrome and high levels of nitric oxide seen in other etiologies of shock, play a role in the genesis and persistence of cardiogenic shock (87,88,89,90,91) (see Fig. 20.2). There is a wide range of EFs demonstrated on patients with cardiogenic shock due to LV failure (average LVEF ∼30%, range <10% to >50%) (92,93,94).
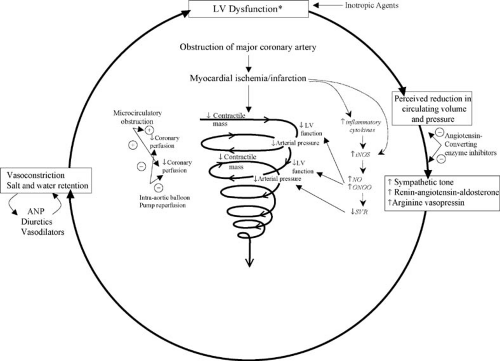 FIGURE 20.2. Pathophysiology of cardiogenic shock caused by primary LV failure. The complex interplay of LV dysfunction, neurohormonal activation, inflammatory mediators, and the vicious cycle of progressively worsening ventricular function that characterizes cardiogenic shock is illustrated. At the onset of shock, SVR is often not elevated, probably because a systemic inflammatory response blunts the expected vasoconstrictor response to hypotension. Later, a neurohumoral response usually increases SVR, as illustrated in the outer circle above. The italicized print represents the newly hypothesized role of the systemic inflammatory response in the genesis and maintenance of shock. For discussion, see text. Abbreviations: ANP, atrial natriuretic peptide; iNOS, inducible nitric oxide synthase; LV, left ventricular; NO, nitric oxide; ONOO, peroxynitrite. (Source: Adapted from Califf RM, Bengtson JR. Cardiogenic shock. N Engl J Med 1994;330:1724–1730 ; andHochman JS. Cardiogenic shock complicating acute myocardial infarction: expanding the paradigm. Circulation 2003;107:2998–3002. ) |
Other Conditions
Conditions that result in high-output failure, including fever and anemia, may occur in acute MI (Table 20.1). Use of negative inotropic agents, such as β-blockers and calcium channel antagonists, particularly in patients with large infarctions, may result in CHF, hypotension, and shock (95). When hypotension is present, noncardiogenic causes of shock, including volume depletion, with or without hemorrhage, sepsis, and PE must be excluded. Even when shock results from primary LV failure, MR caused by papillary muscle dysfunction (96,97) often contributes to shock. Each of the mechanical causes of cardiogenic shock discussed later in this chapter must be excluded. CHF or shock may develop in response to severe emotional stress in the absence of obstructive CAD, as seen in LV Apical Ballooning/Takotsubo Cardiomyopathy (98,99). Aortic dissection with acute aortic regurgitation, tamponade, or both may mimic acute MI with cardiogenic shock (see Table 20.1). An acute MI may occur in association with another catastrophic event (e.g., ruptured aortic aneurysm, ischemic bowel), and the diagnosis may be difficult in an obtunded patient in shock.
Clinical Profile: Congestive Heart Failure and Cardiogenic Shock
The Killip clinical classification system (100) stratifies patients based on clinical evidence of LV failure:
Killip class I is defined as no evidence of CHF.
Killip class II is defined as presence of a third heart sound gallop, basilar rales, or both.
Killip class III is defined as pulmonary edema.
Killip class IV is defined as cardiogenic shock.
TABLE 20.1 Cardiogenic Shock: Differential Diagnosis |
|---|



