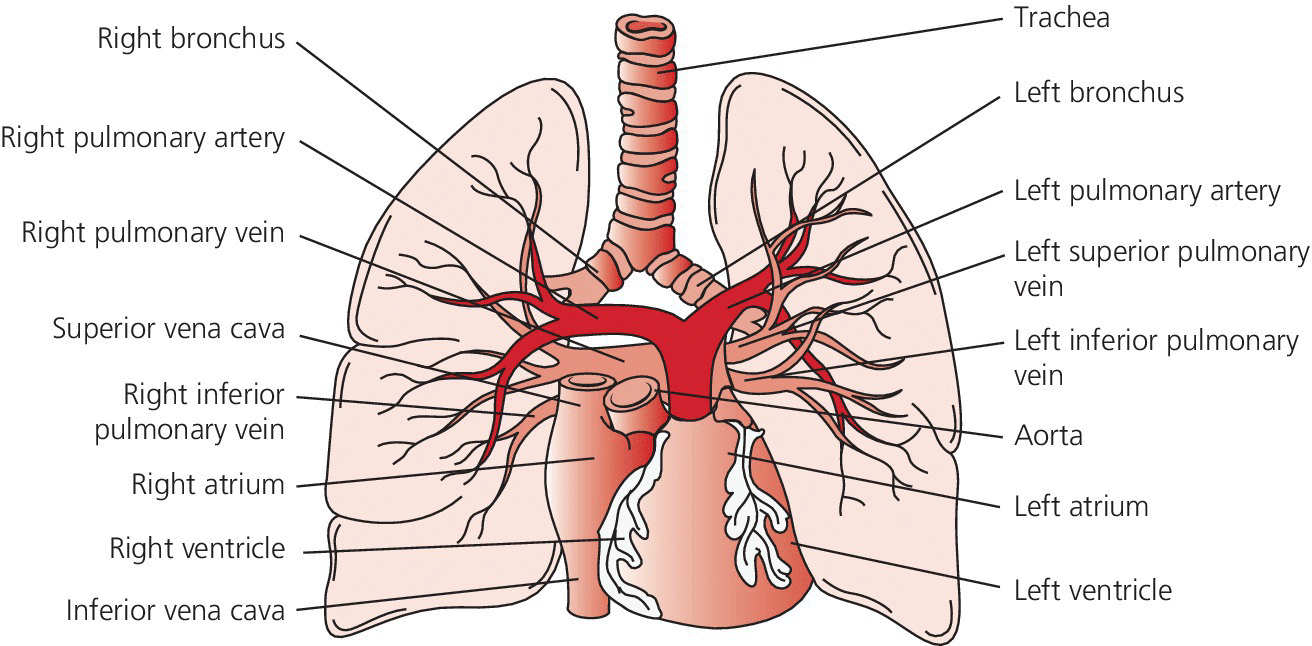
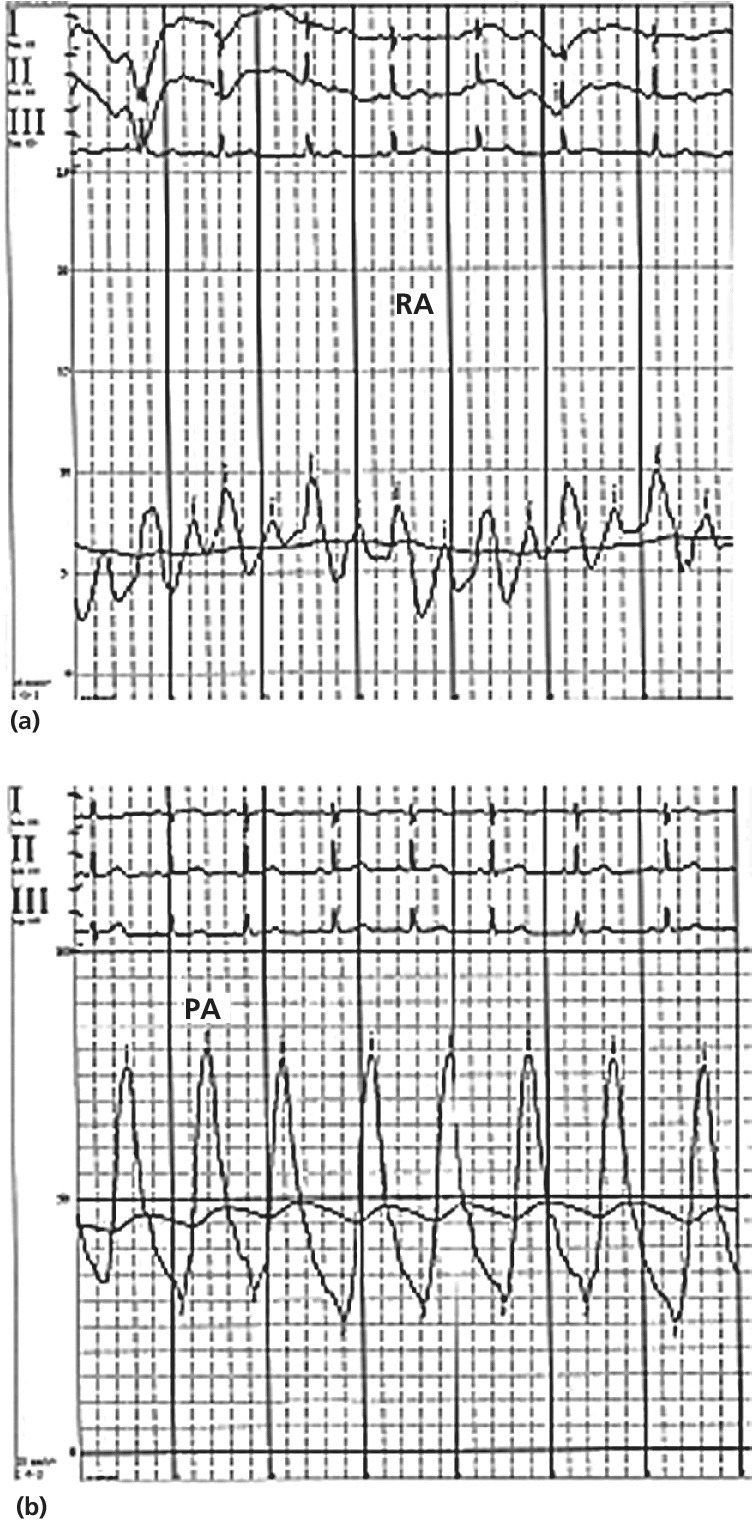
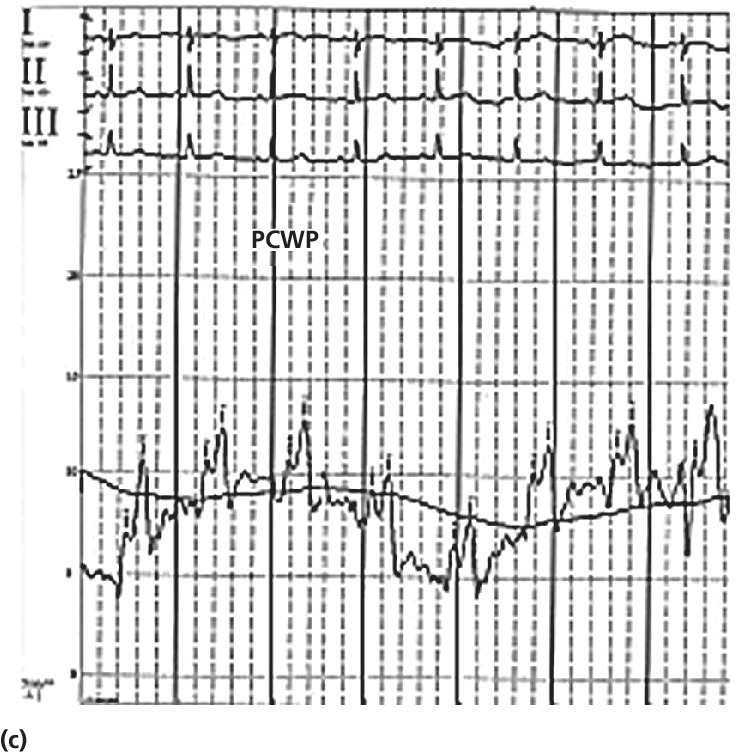
CHAPTER 26 Lisa J. Rose‐Jones, Daniel Fox, David P. McLaughlin and George A. Stouffer The normal pulmonary circulation consists of the pulmonary arteries, which receive deoxygenated blood from the thinly walled, compliant right ventricle. The pulmonary arteries deliver this blood to the capillary beds of the respiratory parenchyma, where carbon dioxide is exchanged for oxygen. Oxygen‐rich blood is then delivered to the left atrium via the pulmonary veins (Figure 26.1). This circulation is characterized by high blood flow, low pressure, and low resistance. The normal adult pulmonary vascular bed is highly distensible and capable of accommodating large increases in blood flow with minimal elevations of pressure. Normal peak systolic pressures in the pulmonary arteries range from 18–25 mm Hg and end‐diastolic pressures in the pulmonary arteries range from 6–10 mm Hg, with a normal mean pressure of less than 20 mm Hg. Figure 26.1 Pulmonary arteries and veins. Pulmonary hypertension is quite simply elevated pressure in the pulmonary arteries and is defined as a mean pulmonary artery pressure (MPAP) ≥25 mm Hg. Right heart catheterization plays a crucial role in diagnosing this disease. It not only helps to determine its presence, but also aids in distinguishing the type and severity. Pulmonary hypertension may be the consequence of increased pulmonary blood flow (e.g., anemia, pregnancy, thyrotoxicosis), increased pulmonary vascular resistance, or elevated left heart pressures. Hemodynamic profiles help to group patients into the World Health Organization (WHO) classification scheme [1]. This allows for patients to be categorized based on similar pathologic findings and guides the clinician as to what therapeutic strategies should be pursued. WHO Group 1 pulmonary arterial hypertension (PAH) is a panvasculopathy of the distal pulmonary arteries that is characterized by medial hypertrophy, intimal proliferation, and fibrosis, with ultimate plexiform lesion formation. There are numerous systemic conditions and heritable genetic mutations that are associated with the development of PAH (Table 26.1). WHO Group 2 pulmonary hypertension due to left heart disease is far and away the most prevalent. Cardiologists will often see this on an almost daily basis. Chronic lung disease and/or hypoxia represent the causative nature in WHO Group 3 pulmonary hypertension. This is felt secondary to hypoxia‐triggered vasoconstriction and vascular remodeling, as well as hyperinflation‐induced compression and obliteration of alveolar vessels. Chronic thromboembolic disease makes up WHO Group 4. It is hemodynamically similar to PAH, but results in an arteriopathy as a consequence of thromboembolic material. While only about 3.8% of patients with acute pulmonary embolic events will develop this at 2 years, it is crucial to identify as it is potentially curable with surgical intervention [2]. Lastly, WHO Group 5 is composed of several different disorders that have not been as well studied. This collection of disorders has unclear and/or multifactorial mechanisms as to the causation of pulmonary hypertension (see Table 26.1). Table 26.1 5th World Health Organization Classification of Pulmonary Hypertension. The hemodynamic hallmark of pulmonary hypertension is elevated pulmonary artery pressures. A mean pulmonary artery pressure (MPAP) ≥25 mm Hg has been well accepted by the pulmonary hypertension community and is used as part of the PAH definition in all major multicenter clinical trials. While it is felt that MPAP of 21–24 mm Hg are likely not normal, much controversy exists on how to label individuals with such readings. Pulmonary artery pressures increase in response to elevated left atrial pressure, cardiac output, or true changes in pulmonary vascular resistance from arterial remodeling. The latter, which represents PAH, has the most evidence base behind targeting management through treatment with pulmonary‐specific vasodilator therapy. Thus, hemodynamics obtained through right heart catheterization are imperative to guide diagnosis and treatment options. Hemodynamic changes in patients with elevated pulmonary pressures depend on the extent of the pulmonary hypertension. The sheer magnitude to which the pulmonary pressures are elevated is important to note; the higher the pressure, the more advanced the disease. The thin‐walled, highly compliant right ventricle can accommodate acute increases in volumes at physiologic pressures (e.g., during exercise), but is not designed to overcome abrupt increases in afterload. Thus, acute RV dilation and failure can accompany sudden increases in pulmonary pressure (e.g., with acute pulmonary embolus). In chronic pulmonary hypertension, RV wall thickness increases (i.e., hypertrophy) to normalize wall stress and myocardial oxygen consumption. However, over time, as pulmonary vascular resistance increases with obliterative pulmonary artery remodeling, the right ventricle reaches a point where it can no longer overcome this afterload. It dilates and starts to fail. As right ventricular failure ensues and forward flow is impaired, the mean pulmonary artery pressure will decrease. It is important not to be fooled by these “lower” than expected MPAP. This end‐stage disease progression is best detected by significant elevations in right atrial pressures (generally >20 mm Hg) associated with a low cardiac index (<2.0 L/min/m2). Both of these have been associated with worse survival [3]. In moderate cases of pulmonary hypertension, it is not uncommon to see very prominent A waves in the right atrial pressure tracings (Figure 26.2). This is a reflection of impaired RV compliance and an increase in RV end‐diastolic pressure (Table 26.2). In more severe cases with significant tricuspid regurgitation, prominent V waves may be present on right atrial pressure tracings. Occasionally with severe cases of RV failure and tricuspid regurgitation, RA pressure tracings take on characteristics similar to ventricular pressure tracings (see Chapter 13). Figure 26.2 Pressure tracings in a patient with pulmonary arterial hypertension. Panel (a) shows right atrial hemodynamic tracing in a patient with portopulmonary PAH. Note the prominent A waves due to a noncompliant, hypertrophied right ventricle. Despite the extremely elevated pulmonary pressures, the patient’s ability to “protect” the right atrial pressure is a good prognostic sign. Panel (b) shows a pulmonary artery pressure waveform on a 100 mm Hg scale. Note the peak systolic pressure of 80 mm Hg and mean pressures of 48–50 mm Hg. Panel (c) shows pulmonary capillary wedge pressure (PCWP) via a balloon‐tipped pulmonary artery catheter. The PCWP is crucial in excluding left heart disease as etiology of elevated pulmonary pressure. It is also necessary for calculating pulmonary vascular resistance, which is now included in the hemodynamic definition of PAH. Table 26.2 Hemodynamic findings in pulmonary hypertension. In addition to MPAP ≥25 mm Hg and pulmonary capillary wedge pressure (PCWP) ≤15 mm Hg, pulmonary vascular resistance >3 Wood units is now included in the definition of PAH from the 5th World Health Symposium on Pulmonary Hypertension in 2013 [4]. Pulmonary vascular resistance (PVR) is calculated by dividing the transpulmonary gradient by the cardiac output:
Pulmonary hypertension
WHO Group 1: Pulmonary arterial hypertension (PAH)
WHO Group 2: Pulmonary hypertension owing to left heart disease
WHO Group 3: Pulmonary hypertension owing to lung diseases and/or hypoxia
WHO Group 4: Chronic thromboembolic pulmonary hypertension (CTEPH)
WHO Group 5: Pulmonary hypertension with unclear and multifactorial mechanisms
Hemodynamic changes associated with pulmonary hypertension
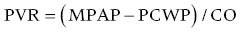
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


