Chapter 7 Wound Healing
Wound-Healing Phases
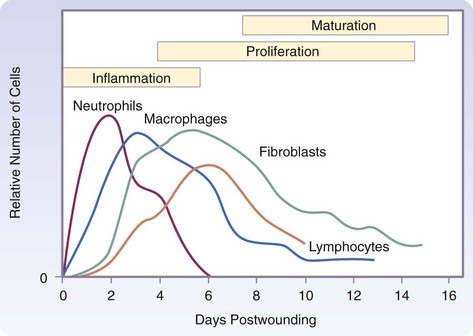
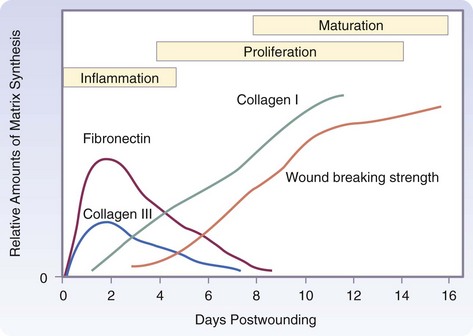
The three phases of wound healing are inflammation, proliferation, and maturation. In a large wound such as a pressure sore, the eschar or fibrinous exudate reflects the inflammatory phase, the granulation tissue is part of the proliferative phase, and the contracting or advancing edge is part of the maturational phase. All three phases may occur simultaneously, and the phases may overlap with their individual processes (Fig. 7-1).
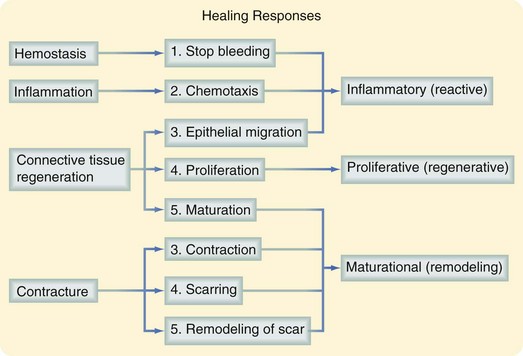
Inflammatory Phase
During the immediate reaction of the tissue to injury, hemostasis and inflammation occur. This phase represents an attempt to limit damage by stopping the bleeding, sealing the surface of the wound, and removing any necrotic tissue, foreign debris, or bacteria present. The inflammatory phase is characterized by increased vascular permeability, migration of cells into the wound by chemotaxis, secretion of cytokines and growth factors into the wound, and activation of the migrating cells (Fig. 7-2).
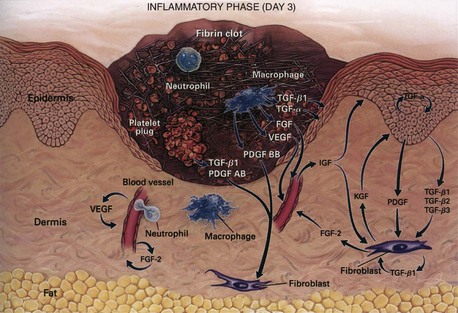
Hemostasis and Inflammation
During an acute tissue injury, blood vessel damage results in initial intense local vasoconstriction of arterioles and capillaries followed by vasodilation and increased vascular permeability (Fig. 7-3). Erythrocytes and platelets adhere to the damaged capillary endothelium, resulting in plugging of capillaries and leading to cessation of hemorrhage. Activation of these platelets by binding to the exposed type IV and V collagen from the damaged endothelium results in platelet aggregation. The initial contact between platelets and collagen requires von Willebrand factor (vWF) VIII, a heterodimeric protein synthesized by megakaryocytes and endothelial cells. Platelet adhesion to the endothelium is primarily mediated through the interaction between high-affinity glycoprotein receptors and the integrin receptor GPIIb-IIIa (αIIbβ3). Platelets also express other integrin receptors that mediate direct binding to collagen (α2β1) and laminin (α6β1) or indirect binding by attaching to subendothelial matrix-bound fibronectin (α5β1), vitronectin (αvβ3), and other ligands.
Chemokines
Macrophage chemoattractant protein (MCP-1, or CCL2) is induced in keratinocytes after injury. It is a potent chemoattractant for monocytes/macrophages, T lymphocytes, and mast cells.1 Expression of this chemokine is sustained in chronic wounds and results in the prolonged presence of polymorphonuclear cells (PMNs) and macrophages, leading to the prolonged inflammatory response.2 CXCL1 (GRO-α) is a potent PMN chemotactic regulator and is increased in acute wounds. It is also involved in reepithelialization.3 Interleukin-8 (IL-8, or CXCL8) expression is increased in acute and chronic wounds.4 It is involved in reepithelialization and induces the leukocyte expression of matrix metalloproteinases (MMPs), which stimulates remodeling. It is also a strong chemoattractant for PMNs and participates in inflammation.4 Relatively low levels of IL-8 are found in fetal wounds and may be why fetal wounds have decreased inflammation and heal without scars.5 Expression of the keratinocyte-produced chemokine interferon inducible protein 10 (IP-10 or CXCL10) is elevated in acute wounds as well as chronic inflammatory conditions.6 It impairs wound healing by increasing inflammation and recruiting lymphocytes to the wound. It also inhibits proliferation by decreasing reepithelialization and angiogenesis and preventing fibroblast migration.3 Stromal cell-derived factor-1 (SDF-1, or CXCL12) is expressed by endothelial cells, myofibroblasts, and keratinocytes and is involved in inflammation by recruiting lymphocytes to the wound and promoting angiogenesis. It is a potent chemoattractant for endothelial cells and bone marrow progenitors from the circulation to peripheral tissues.7 It also enhances keratinocyte proliferation, resulting in reepithelialization.8
Polymorphonuclear Cells
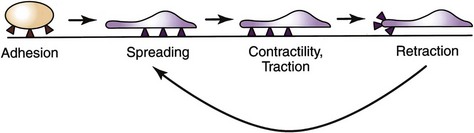
Integrins are crucial for cell motility and are required in inflammation and normal wound healing, as well as in embryonic development and tumor metastases. After extravasation, PMNs, attracted by chemotaxins, migrate through the ECM by means of transient interactions between integrin receptors and their ligands. Four phases of integrin-mediated cell motility have been described: adhesion, spreading, contractility or traction, and retraction. Activation of specific integrins though ligand binding has been shown to increase cell adhesion and activate reorganization of the cell’s actin cytoskeleton. Spreading is characterized by the development of lamellipodia and filopodia. Traction at the leading edge of the cell develops through binding of integrin, followed by translocation of the cell over the adherent segment of the plasma membrane. The integrin is shifted to the rear of the cell and releases its substrate, thereby permitting cell advancement (Fig. 7-4). Regulation of integrin function by adhesive substrates offers a mechanism for local control of migrant cells. Within the assembled framework of the ECM, binding sites for integrins have been identified on collagen, laminin, and fibronectin.
Macrophages
The macrophage is the one cell that is truly crucial to wound healing in that it serves to orchestrate the release of cytokines and stimulate many of the subsequent processes of wound healing (Fig. 7-5). Macrophages in the wound appear at the same time that neutrophils disappear. Macrophages induce apoptosis of PMNs. Chemotaxis of migrating blood monocytes occurs within 24 to 48 hours. Chemotactic factors specific for monocytes include bacterial products, complement degradation products (C5a), thrombin, fibronectin, collagen, TGF-β, and PDGF-BB. Monocyte chemotaxis is also facilitated by the interaction of integrin receptors on the monocyte surface with ECM proteins such as fibrin and fibronectin. The β integrin receptor also transduces the signal for macrophage phagocytic activity. Activated integrin expression promotes adhesion-mediated gene induction in monocytes that transforms them into wound macrophages; such transformation results in increased phagocytic activity and selective expression of cytokines and signal transduction elements by messenger RNA (mRNA), including the early growth response genes EGR2 and c-fos. Macrophages have specific receptors for immunoglobulin G (IgG; Fc receptor), C3b (CR1 and CR3), and fibronectin (integrin receptors) that permit surface recognition of opsonized pathogens and facilitate phagocytosis.
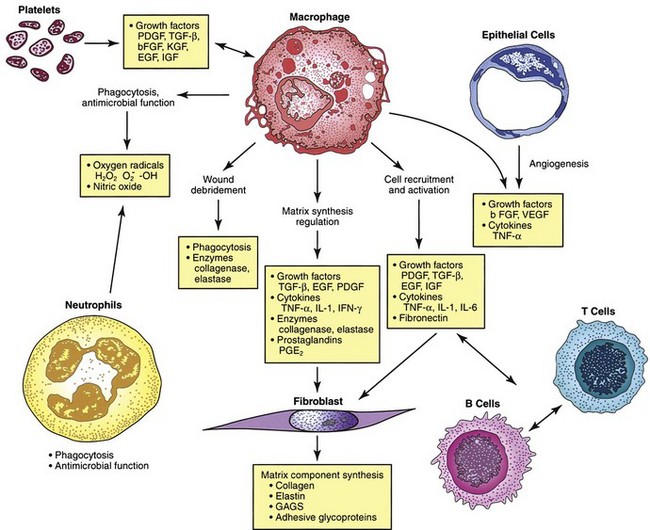
Wound macrophages release proteinases, including matrix metalloproteinases (MMP-1, MMP-2, MMP-3, and MMP-9; Fig. 7-6), that degrade the ECM and are crucial for removing foreign material, promoting cell movement through tissue spaces, and regulating ECM turnover. This activity is dependent on the cAMP pathway and thus can be blocked by nonsteroidal anti-inflammatory drtugs (NSAIDs) or glucocorticoid drugs. Colchicine and retinoic acid appear to decrease collagenase production as well.
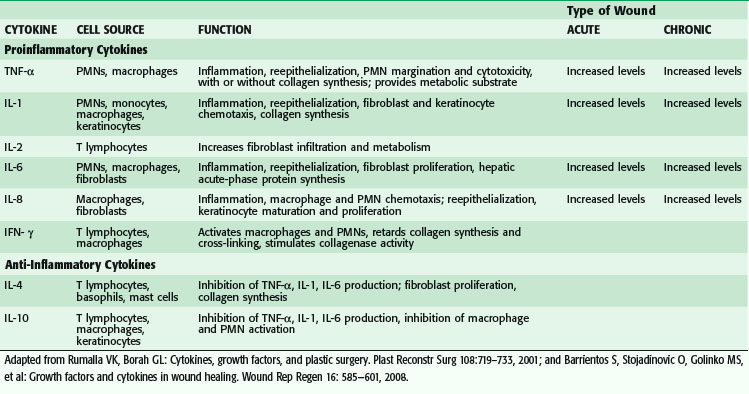
Macrophages secrete numerous cytokines and growth factors (Tables 7-1 and 7-2). IL-1, a proinflammatory cytokine, is an acute-phase response cytokine. This endogenous pyrogen causes lymphocyte activation and stimulation of the hypothalamus, thereby inducing the febrile response. It also directly affects hemostasis by inducing the release of vasodilators and stimulating coagulation. Its effect is further amplified as endothelial cells produce it in the presence of TNF-α and endotoxin. IL-1 has numerous effects, such as enhancement of collagenase production, stimulation of cartilage degradation and bone reabsorption, activation of neutrophils, regulation of adhesion molecules, and promotion of chemotaxis. It stimulates other cells to secrete proinflammatory cytokines. Its effects also extend into the proliferative phase, during which it increases fibroblast and keratinocyte growth and collagen synthesis. Studies have demonstrated increased levels of IL-1 in chronic nonhealing wounds, thus suggesting its role in the pathogenesis of poor wound healing. The early beneficial responses of IL-1 in wound healing appear to be maladaptive if elevated levels last beyond the first week after injury.
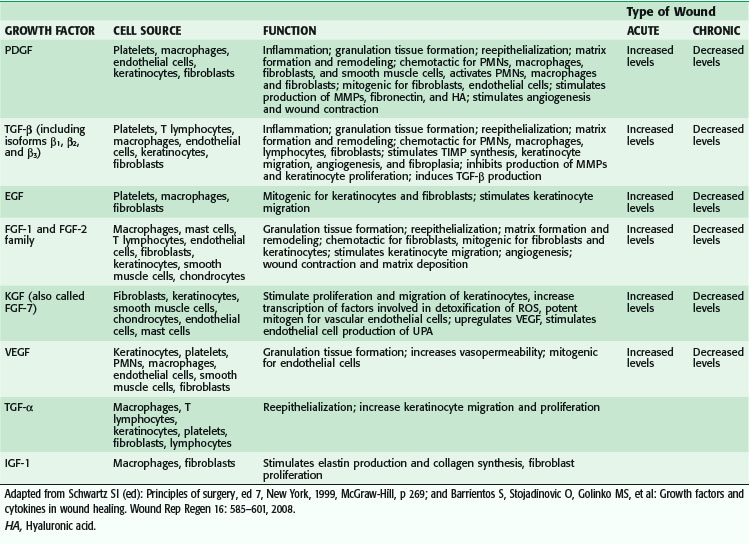
Macrophages also release growth factors that stimulate fibroblast, endothelial cell, and keratinocyte proliferation and are important in the proliferative phase (see Table 7-2). Macrophage-secreted PDGF stimulates collagen and proteoglycan synthesis. PDGF exists as three isomers—PDGF-AA, PDGF-AB, and PDGF-BB. However, the PDGF-BB isomer is the only growth factor preparation approved by the U.S. Food and Drug Administration and is the most widely studied clinically. Topical application of recombinant PDGF has improved wound-breaking strength and healing time in human and murine models of acute wounding. Administration of PDGF-BB has improved wound closure in chronic and diabetic nonhealing ulcers in humans and rodents but did not have the same effect in steroid-treated animals.
Proliferative Phase
Angiogenesis
VEGF, a member of the PDGF family of growth factors, has potent angiogenic activity. It is produced in large amounts by keratinocytes, macrophages, endothelial cells, platelets, and fibroblasts during wound healing.9–12 Cell disruption and hypoxia, hallmarks of tissue injury, appear to be strong initial inducers of potent angiogenic factors at the wound site, such as VEGF and its receptor. VEGF family members include VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor (PLGF).13 VEGF-A promotes early events in angiogenesis and subsequently is crucial to wound healing.14 It binds to tyrosine kinase surface receptors Flt-1 (VEGF receptor-1, or VEGFR-1) and KDR (VEGF receptor-2, or VEGFR-2).15 Flt-1 is required for blood vessel organization, whereas KDR is important for endothelial cell chemotaxis, proliferation, and differentiation.16,17 Animal studies have shown that VEGF-A administration restores impaired angiogenesis found in diabetic ischemic limbs18; however, other studies have shown that exogenous VEGF results in vascular leakage and disorganized blood vessel formation.19,20 VEGF-C, which is also elevated during wound healing, is primarily released by macrophages and is important during the inflammatory phase of wound healing.21 Although it works primarily through VEGF receptor-3 (VEGFR-3), which is expressed in macrophages and lymphatic endothelium, it can also activate VEGFR-2, thereby increasing vascular permeability.21 In vivo administration of VEGF-C in an animal model using an adenoviral vector to genetically diabetic mice resulted in accelerated healing.13 PLGF is another proangiogenic factor that is elevated after wounding. It is involved in inflammation and expressed by keratinocytes and endothelial cells. It is believed to work synergistically with VEGF, thereby potentiating its proangiogenic function.22
Epithelialization
Extracellular Matrix
In connective tissue, proteoglycan molecules form a gel-like ground substance. This highly hydrated gel allows the matrix to withstand compressive force while permitting rapid diffusion of nutrients, metabolites, and hormones between blood and tissue cells. Collagen fibers within the matrix serve to organize and strengthen it, whereas elastin fibers give it resilience and matrix proteins have adhesive functions.23
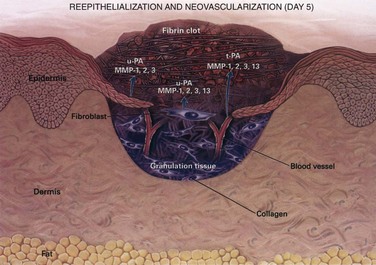
The wound matrix accumulates and changes in composition as healing progresses, balanced between new deposition and degradation (Fig. 7-7). The provisional matrix is a scaffold for cellular migration and is composed of fibrin, fibrinogen, fibronectin, and vitronectin. GAGs and proteoglycans are synthesized next and support further matrix deposition and remodeling. Collagens, which are the predominant scar proteins, are the end result. Attachment proteins, such as fibrin and fibronectin, provide linkage to the ECM through binding to cell surface integrin receptors.
Collagen Structure
Collagens are found in all multicellular animals and are secreted by a variety of cell types. They are a major component of skin and bone and constitute 25% of the total protein mass in mammals. The proline- and glycine-rich collagen molecule is a long, stiff, triple-stranded helical structure that consists of three collagen polypeptide α chains wound around one another in a ropelike superhelix. With its ringlike structure, proline provides stability to the helical conformation in each α chain, whereas glycine, because of its small size, allows tight packing of the three α chains to form the final superhelix. There are at least 20 types of collagen, the main constituents of connective tissue being types I, II, III, V, and XI. Type I is the principal collagen of skin and bone and is the most common.23 In adults, the skin is approximately 80% type I and 20% type III. In newborns, the content of type III collagen is greater than that found in adults. In early wound healing, there is also increased expression of type III collagen. Type I collagens are the fibrillar, or fibril-forming, collagens. They are secreted into the extracellular space, where they assemble into collagen fibrils (10 to 300 nm in diameter), which then aggregate into larger, cable-like bundles called collagen fibers (several micrometers in diameter).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


