Chapter 57 The Spleen
Splenic Anatomy
The spleen is an 80- to 300-g organ that initially develops from mesenchymal cells in the dorsal mesogastrium during week 5 of embryogenesis and settles into the left uppermost aspect of the abdomen. Its superior surface is roofed by the diaphragm, separating it from the pleura. It should be noted, however, that the costodiaphragmatic recess extends to the inferiormost aspect of a normal-sized spleen. The spleen’s visceral relationships include the greater curvature of the stomach, splenic flexure of the colon, apex of the left kidney, and tail of the pancreas (Fig. 57-1). It is protected by ribs 9, 10, and 11and is suspended in its location by multiple peritoneal reflections, the splenophrenic, gastrosplenic, splenorenal, and splenocolic ligaments. In patients without portal hypertension, the splenophrenic and splenocolic ligaments are relatively avascular. The gastrosplenic ligament carries the short gastric vessels in its superior aspect and the left gastroepiploic in its inferior aspect. The splenorenal ligament houses the splenic artery and vein, as well as the tail of the pancreas. The tail of the pancreas abuts the splenic hilum in 30% of individuals and is within 1 cm of the hilum in 70%.
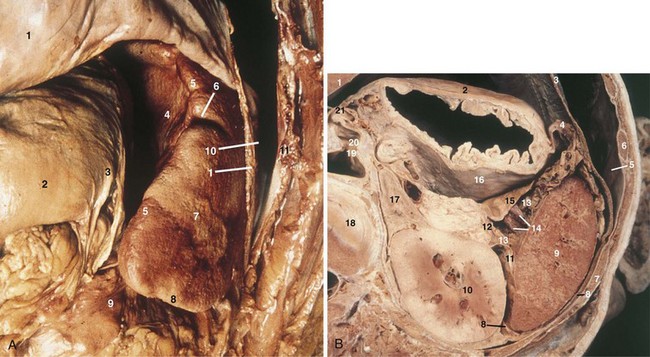
The splenic artery, a branch of the celiac trunk, is a tortuous vessel that gives off multiple branches to the pancreas as it travels along its posterior aspect (Fig. 57-2). There are two typical arrays of the splenic artery—the magistral, which branches into terminal and polar arteries near the hilum of the spleen and the distributed, which, as the name implies, gives off its branches early and distant from the hilum. There is typically a superior polar artery, which sometimes communicates with the short gastric arteries, superior, middle, and inferior terminal arteries, and an inferior polar artery. Knowing these variable distributions is necessary when performing resections, especially a spleen-preserving procedure. Because of the variable nature of the splenic artery, one must be cautious when operating near this vessel and its tributaries.
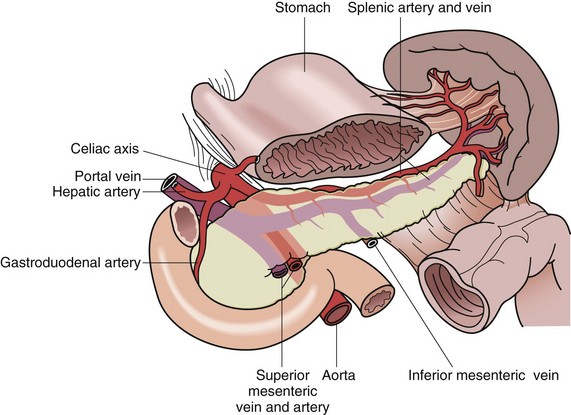
FIGURE 57-2 Anatomic relationships of the splenic vasculature.
(From Economou SG, Economou TS: Atlas of surgical techniques, Philadelphia, 1966, WB Saunders, p 562.)
Splenic Function
During fetal development, the spleen has important hematopoietic functions, which include white and red blood cell production. This production is usurped by the bone marrow by the fifth month of gestation and, under normal conditions, the spleen has no significant hematopoietic function beyond this point. In certain pathologic conditions, such as myelodysplasia, the spleen may reacquire this function. Beyond hematopoiesis, the specialized vasculature in the spleen is directly related to its remaining functions, defense and cleansing. It is likely that the spleen’s mechanical filtration contributes to control of infection by removing pathogens within cells (e.g., malaria) or circulating in the plasma. This filtration may be particularly important for removing microorganisms for which the host does not have a specific antibody (Box 57-1).
Box 57-1 Adapted from Eichner ER: Splenic function: Normal, too much and too little. Am J Med 66:311–320, 1979.
Biologic Substances Removed by the Spleen
The immune functions of the spleen become obvious after splenectomy, when patients are noted to be significantly at risk for infection. The most serious sequela is overwhelming postsplenectomy infection (OPSI), with meningitis, pneumonia, or bacteremia.1 Older studies have demonstrated that the risk of OPSI is greatest within the first 2 years after splenectomy but recent studies have confirmed that a lifelong risk remains. One third of cases occur more than 5 years after surgery, with the overall incidence reported to be 3.2% to 3.5%. For those who acquire OPSI, mortality is between 40% and 50%.2 The risk is greatest in patients with thalassemia major and sickle cell disease. OPSI is typically caused by polysaccharide-encapsulated organisms, such as Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae. These and other organisms are identified and bound by antibodies and complement components in preparation for phagocytosis by macrophages in the spleen. After splenectomy, the antibodies continue to bind but digestion by splenic macrophages is no longer possible.
Asplenic patients have been noted to express similar postvaccination immunoglobulin G (IgG) antibody levels when comparing timing of pneumococcal vaccinations in postsplenectomy trauma patients; functional antibody levels, however, were lower.3 Also, asplenic patients have been found to express subnormal IgM levels and their peripheral blood mononuclear cells exhibit a suppressed immunoglobulin response. The risk of developing OPSI or asplenic or hyposplenic overwhelming sepsis for reasons other than surgical removal of the spleen is linked to patient understanding of the risks of infection.4 Registries that allow for long-term follow-up and periodic teaching of current recommendations should be considered for this high-risk population.5,6
The filtration consists of two methods of blood flow within the spleen, the closed and open systems. In the closed system, blood flows directly from arteries to veins. In the open system, most of the spleen’s blood flow occurs when blood flows through the arterioles and then trickles through a sievelike parenchyma made up of reticuloendothelial cells into the splenic sinuses before draining into the venous system (Fig. 57-3). The cellular elements are directed toward these reticuloendothelial cells, in which cellular cleansing processes take place. These include removal of senescent cells, cellular inclusion (e.g., red cell nucleoli), parasites, and sequestration of red cells (for maturation) and platelets (reservoir). The plasma is directed to the lymphoid tissue, where soluble antigens stimulate the production of antibodies.
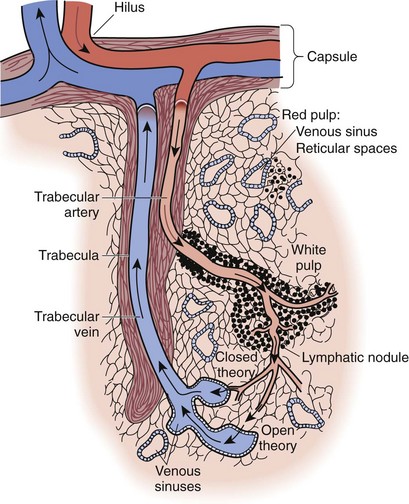
FIGURE 57-3 Structure of the sinusoidal spleen showing the open and closed blood flow routes.
(From Bellanti JA: Immunology: Basic brocesses. Philadelphia, 1979, WB Saunders.)
Splenectomy
Splenectomy may be performed for a number of reasons and conditions.
Benign Hematologic Conditions
Immune Thrombocytopenic Purpura
The typical presentation of ITP is characterized by purpura, epistaxis, and gingival bleeding. Less commonly, gastrointestinal bleeding and hematuria are noted. Intracerebral hemorrhage is a rare but sometimes fatal presentation. The diagnosis of ITP involves the exclusion of other relatively common causes of thrombocytopenia—pregnancy, drug-induced thrombocytopenia (e.g., heparin, quinidine, quinine, sulfonamides), viral infections, and hypersplenism (Box 57-2). Mild thrombocytopenia may be seen in approximately 6% to 8% of otherwise normal pregnancies and in up to 25% of women with preeclampsia. Drug-induced thrombocytopenia is thought to occur rarely, in approximately 20 to 40 cases/million users of common medications, such as trimethoprim-sulfonamide and quinine. Other medications, such as gold salts, have a higher incidence, almost 1% of users.7 Viral infection (e.g., hepatitis C virus [HCV], HIV, rarely, Epstein-Barr virus [EBV]) can be responsible for thrombocytopenia independent of splenic sequestration. Once again, other processes must be ruled out but health care providers can be confident of these causative factors if platelet counts improve with successful treatment of the responsible infection. Bacterial infection, specifically Helicobacter pylori, has also been linked to infection-related thrombocytopenia that improves with eradication. Other causes are listed in Box 57-2; spurious laboratory values caused by platelet clumping or the presence of giant platelets should not be ignored.
Box 57-2 Adapted from George JN, El-Harake MA, Raskob GE: Chronic idiopathic thrombocytopenic purpura. N Engl J Med 331:1207–1211, 1994.
Differential Diagnosis of Immune Thrombocytopenic Purpura
Common Causes of Thrombocytopenia
Pregnancy (gestational thrombocytopenia, preeclampsia)
Drug-induced thrombocytopenia (common drugs include heparin, quinidine, quinine, sulfonamides)
Viral infections, such as HIV, rubella, infectious mononucleosis
Other Causes of Thrombocytopenia Mistaken for Immune Thrombocytopenic Purpura
Thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome
Management of ITP depends primarily on the severity of the thrombocytopenia.8 Asymptomatic patients with platelet counts higher than 50,000/mm3 may be observed without further intervention. Platelet counts of 50,000/mm3 and higher are rarely associated with clinical sequelae, even with invasive procedures. Patients with slightly lower platelet counts, between 30,000 and 50,000/mm3, may always be observed but with more routine follow-up because they are at increased risk for progressing to severe thrombocytopenia. Initial medical treatment for patients with platelets counts less than 50,000/mm3 and symptoms such as mucous membrane bleeding, high-risk conditions (e.g., active lifestyle, hypertension, peptic ulcer disease), or platelet counts less than 20,000 to 30,000/mm3, even without symptoms, is glucocorticoid administration (typically, prednisone, 1 mg/kg body weight/day). Clinical response with increases in platelet levels to higher than 50,000/mm3 is seen in up to two thirds of patients within 1 to 3 weeks of initiating treatment. Of patients treated with steroids, 25% will experience a complete response. Patients with platelet counts higher than 20,000/mm3 who remain symptom-free, or who experience minor purpura as their only symptom, do not require hospitalization. Hospitalization may be required for patients whose platelets counts remain below 20,000/mm3 with significant mucous membrane bleeding and is required for those who have life-threatening hemorrhage. Platelet transfusion is indicated only for those who experience severe hemorrhage. IV immunoglobulin is important for the treatment of acute bleeding, in pregnancy, or for patients being prepared for operation, including splenectomy. The usual dose is 1 g/kg body weight/day for 2 days. This dose usually increases the platelet count within 3 days; it also increases the efficacy of platelet transfusions.
Prior to the establishment of glucocorticoids as treatment for ITP in 1950, splenectomy was the treatment of choice.8 For those two thirds of patients in whom glucocorticoids result in the normalization of platelet counts, no further treatment is necessary. For patients with severe thrombocytopenia, with counts less than 10,000/mm3 for 6 weeks or longer, those with thrombocytopenia refractory to glucocorticoid treatment, or those who require toxic doses of steroid to achieve remission, the treatment of choice is to proceed to splenectomy. Splenectomy is also the treatment of choice for patients with incomplete response to glucocorticoid treatment and for pregnant women in the second trimester of pregnancy who have also failed steroid treatment or IV Ig therapy with platelet counts less than 10,000/mm3 without symptoms or less than 30,000/mm3 with bleeding problems. It is not necessary to proceed to splenectomy for patients who have platelet counts higher than 50,000/mm3, have had ITP for longer than 6 months, are not experiencing bleeding symptoms, and who are not engaged in high-risk activities. A recent review of short-term and long-term failure of laparoscopic splenectomy has reported an overall approximate failure rate of 28% at 5 years after splenectomy.9
A systematic review of 436 published articles from 1966 to 2004 has reported that 72% of patients with ITP had a complete response to splenectomy. Relapse occurred in a median of 15% of patients (range, 1% to 51%), with a median follow-up of 33 months.10
In addition to relapse rates, predictors of successful splenectomy were examined. Of the variables in the multivariate model, age at the time of splenectomy was an independent variable that was most correlated with response.10 Younger patients had improved responses. Preoperative indium-111 (111In)–labeled platelet scintigraphy with platelets sequestered predominantly within the spleen had a significantly higher response rate than those noted to have hepatic sequestration.11
Most patients will exhibit improved platelet counts within 10 days postoperatively and durable platelet responses are associated with patients who have platelet counts of 150,000/mm3 by postoperative day 3 or more than 500,000/mm3 by the postoperative day 10. Even with splenectomy, however, some patients may relapse (~12%; range, 4% to 25%).12 A recent review of 1223 ITP patients has estimated the long-term failure rate of laparoscopic splenectomy at approximately 8% and approximately 44/1000 patient-years of follow-up.9 Another study has estimated the complete response of ITP patients postsplenectomy to be 66%.10
Although a thorough search for accessory spleens is completed during the initial surgery, evaluation for a missed accessory spleen must be undertaken in patients who experience a relapse. In their evaluation of 394 patients treated with laparoscopic splenectomy, Katkhouda and colleagues12 noted 15% of patients with accessory spleens. In those with accessory spleens, examination of a peripheral blood smear will lack the characteristic red cell morphology resulting from excision of the spleen. Radionuclide imaging may also be helpful in locating the presence and location of any accessory splenic tissue. Patients with chronic ITP in whom an accessory spleen is identified should have this removed, as long as the patient can withstand the surgical risk.
Other treatment options for these patients include observation of stable nonbleeding patients with platelet counts higher than 30,000/mm3, long-term glucocorticoid therapy, and treatment with azathioprine or cyclophosphamide. Recent evidence regarding thrombopoietin receptor agonists may offer a novel medical therapy for patients with no response to steroids, IV immunoglobulin therapy, or splenectomy.13
Approximately 10% to 20% of otherwise asymptomatic patients with HIV will develop ITP. Splenectomy is a safe treatment option for this cohort of patients and may actually delay HIV disease progression.14,15
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


