Chapter 40 Endocrine Pancreas
The pancreas is a digestive organ located in the retroperitoneum (Fig. 40-1) that has endocrine and exocrine function. The endocrine cells are organized in discrete clumps throughout the pancreas, called islets of Langerhans. The primary physiologic function of the endocrine pancreas can be summarized as regulation of body energy, largely through hormonal control of carbohydrate metabolism. The islets secrete hormones directly into the bloodstream in endocrine fashion. Insulin is the hormone of energy storage, while glucagon is the hormone of energy release. Additional pancreatic endocrine hormones, such as somatostatin, play a role in the complex regulation of pancreatic exocrine secretion and digestion.
The pancreas was first identified by a Greek anatomist and surgeon, Herophilus (335-280 BC). In medieval Persia, in 1025, Avicenna provided the first detailed account of diabetes mellitus in the Canon of Medicine. He described a patient with an abnormal appetite, collapse of sexual function, and the sweet taste of diabetic urine. In 1889, Minkowski and von Mering, who were studying fat absorption in dogs after pancreatectomy, noted that the urine attracted flies. On analysis of the urine, they documented glucosuria and ketonuria. They also noted that surgical removal of the pancreas led to eventual coma and death. In 1869, while a medical student, Paul Langerhans described collections of pale staining cells within the pancreas, the islets that now bear his name (see Fig. 40-1). Eugene Opie was the first to associate diabetes with microscopic hyaline changes in the islets of Langerhans. Frederick Banting and Charles Best in Toronto discovered insulin in 1922. Banting and Best surgically ligated the pancreas of one set of dogs, leading to atrophy of the exocrine pancreas. They then removed and homogenized the pancreas and injected the homogenized extract into a diabetic dog, temporarily reversing this condition; a few injections each day could keep it healthy and free of symptoms. Banting and Best were awarded a Nobel Prize for this work.
Adult human pancreatic islets contain multiple cell types (Table 40-1). Alpha (A) cells secrete glucagon, beta (B) cells secrete insulin, delta (D) cells secrete somatostatin and vasoactive intestinal peptide (VIP), and F cells secrete pancreatic polypeptide (PP). Gastrin-producing cells are normally present in the fetal pancreas only. Islet cell tumors may secrete one or more of these hormones. The resulting syndromes are named for the peptide whose clinical symptoms predominate.
Histomorphology of Islets
The islet cell types are not distributed evenly within the islets. B cells constitute approximately 70% of the islet cell mass and are located centrally within the islet.1 Insulin is the main secretory product of B cells, but they have also been shown to secrete amylin and cholecystokinin (CCK; see Table 40-1).2 The A cells, located in the periphery, secrete glucagon and constitute approximately 10% of the islet cell mass. The D cells are evenly distributed throughout the islet and constitute approximately 5% of the islet cell mass. D cells secrete somatostatin and D2 cells secrete VIP. Also located peripherally, F cells secrete PP. B and D cells are concentrated in the body and tail of the pancreas and F cells are concentrated in the head and uncinate process. This distribution is important clinically, because resection of different parts of the pancreas will have varying endocrine effects.
Pancreatic islets have a rich portal microcirculation that has significance in endocrine to endocrine cell signaling. Afferent arterioles enter the islet in an area of discontinuity in the peripheral, non-B cell mantle of cells. The order of islet cellular perfusion and interaction is from the B cell core outward to the mantle, and the mantle is further subordered with most D cells downstream or distal to most A cells. This allows B cells to inhibit A cell secretion and A cells to stimulate D cell secretion.3
Embryology of the Endocrine Pancreas
Islet cells were initially believed to arise from neural crest cells. Gittes and Rutter4 have studied the patterns of expression of hormonal messenger RNA and concluded that both endocrine and exocrine cells of the pancreas arise from the foregut endoderm, a view that is now generally accepted. Early glucagon-positive endocrine cells convert to nonepithelial cells and lose connection with the lumen and tight junctions. This conversion to a nonepithelial location of endocrine cells has been postulated to entail a change in cell division polarity, from perpendicular to the basement membrane to parallel to the basement membrane. There also appears to be downregulation of pdx1, a key marker of early pancreatic progenitor cells, in these endocrine progenitor cells as they become nonepithelial. This conversion process has been postulated to parallel mesenchymal to mesenchyme transformation.
Endocrine Physiology
Insulin
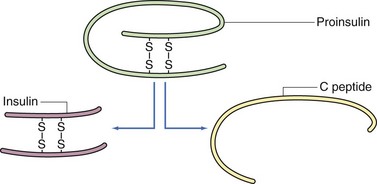
Insulin is a 56–amino acid polypeptide with a molecular weight of 6 kDa. It consists of two polypeptide chains (A and B) joined by two disulfide bridges. Although the amino acid sequence varies among species, the locations of the disulfide bridges are highly conserved and are critical for its biologic activity. Insulin is synthesized as a precursor peptide, called proinsulin; the two polypeptide chains are joined by means of connecting peptide (C peptide). In response to pancreatic B cell stimulation by glucose, proinsulin is synthesized in the endoplasmic reticulum and transported to the Golgi complex, where it is cleaved into insulin and the residual C peptide (Fig. 40-2). Insulin is then moved via microtubules into secretory granules, where it is released directly into the bloodstream via exocytosis. C peptide and insulin are secreted in equimolar amounts.
There is significant secretory reserve of insulin within the pancreas. Destruction or removal of 80% of the pancreatic islet cell mass is necessary before endocrine dysfunction becomes clinically apparent in the form of type 1 (insulin-dependent) diabetes. Defects in the synthesis and cleavage of insulin can lead to rare forms of diabetes mellitus, such as Wakayama syndrome and proinsulin syndrome.5
Surgical Treatment of Diabetes
Autologous Islet Cell Transplantation
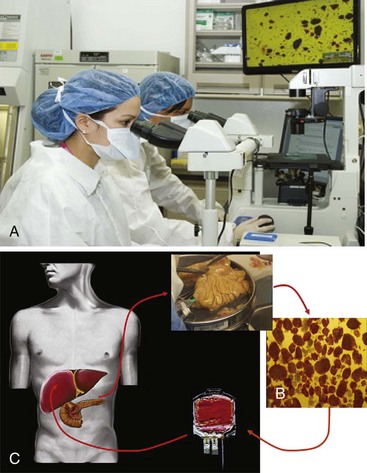
Total or partial pancreatectomy with islet autotransplantation is being offered in several centers in the United States. This option has the potential to treat the symptoms of chronic pancreatitis definitively while preventing the onset of diabetes in certain patients. Other patients remain or become insulin-dependent but retain significant insulin and glucagon secretion and the benefits of endogenous C peptide production, thus making the resulting diabetes easier to control. Our dedicated islet isolation facility and a diagrammatic representation of the process are shown in Figure 40-3. Patients undergo pancreatectomy; the pancreatic tissue is immediately digested with the use of enzyme solutions containing collagenase and neutral proteases and the islet cells are purified. The islet cells are then returned to the patient via infusion into the portal vein. The islet cells engraft in the liver and produce insulin and C peptide and glucose levels are measured to evaluate the function of the transplanted islets.
Depending on center expertise and experience, variable results following pancreatic islet autotransplantation have been reported. Insulin independence is as high as 40% to 50% initially in some patients,6–8 but there is a notable decline in islet function over time, with increased insulin requirements in the 10 years following transplantation, with only about 10% of patients remaining insulin-independent. Although insulin-independence is not always achieved, most patients are C peptide positive and have diabetes that is more manageable. In addition, all studies have demonstrated improvement in pain and other symptoms of chronic pancreatitis.9,10 Success rates depend on the number of isolated and transplanted islets and transplanted islets as well as the cause of the pancreatic disease. Patients who are not diabetic before autotransplantation and younger patients (especially preadolescents) achieve the best results. The most feared procedure-related complication is thrombosis of the portal vein, occurring in less than 1% of cases.
Immune Therapy, Pancreatic Transplantation, and Islet Allotransplantation
Type 1 diabetes results from autoimmune destruction of pancreatic islets. Immune treatment for type 1 diabetes is currently being investigated. There is a growing body of evidence to suggest that the autoimmunity observed in patients with type 1 diabetes is the result of an imbalance between autoaggressive and regulatory T cell subsets.11,12 Vaccination with selected T cell receptor autoantigens has been shown to generate autoantibodies and the autoaggressive T cell clones, which are reacting to beta cells. The induction of a lasting, robust immune response generating autoantigen-specific regulatory T cells provides strong justification for further testing of this therapy for type 1 diabetes.
The current surgical treatment for type 1 diabetes is transplantation of allogeneic islet cell tissue by means of whole organ transplantation or transplantation of isolated islets, usually infused into the portal vein. Pancreatic transplantation was first performed in 1966 by Kelly and colleagues. From 1966 through 2008, over 30,000 pancreas transplantations have been reported to the International Pancreas Transplant Registry, including more than 22,000 from the United States.13 Whole organ pancreas transplants restore euglycemia almost immediately following transplantation, and 1-year graft survival rates in the United States have improved to 85% for simultaneous pancreas-kidney transplants, 78% for pancreas after kidney transplants, and 76% for pancreas-only transplants. Recipients experience immediate normal fasting and postprandial glucose levels, and hemoglobin A1c levels return to normal. With the observed decrease in morbidity and mortality, recipients who become insulin-independent report a better quality of life, despite the need for immunosuppression. They also experience stabilization or improvements in retinopathy, nephropathy, neuropathy, and microvascular and macrovascular diseases normally associated with poor glucose control. Therefore, for the select group of patients with labile diabetes who do not respond well to conventional approaches or insulin pumps, whole pancreas transplantation remains the gold standard for treatment.
Allogeneic islet cell transplantation is the other option. Currently, long-term insulin independence remains elusive for patients undergoing allogeneic islet transplantation. The data show that even with patients who receive multiple infusions, few remain normoglycemic over time. Data from the Collaborative Islet Transplant Registry (CITR) have demonstrated that 70% of patients achieve insulin independence within the first year (including patients with multiple infusions) but, by the third year, the percentage of patients who remain euglycemic is closer to 35%.13a The partial pancreatic endocrine function confers some benefit, with decreased occurrence of severe hypoglycemic events, abatement of hypoglycemic unawareness, persistent C peptide levels, improvement in glycemic control, and stabilization of diabetic complications.
Whole pancreas transplantation procedures currently outnumber islet transplantation procedures. Pancreas transplantation is associated with a higher surgical morbidity, whereas islet transplantation is a less invasive method of achieving insulin independence. However, pancreas transplantation is associated with a higher success rate. In addition, islet cell transplantation requires two donors per recipient to maintain graft function.14 Stem cell therapy offers the potential of producing an unlimited source of cells, and a growing number of studies have demonstrated successful in vitro differentiation and expansion of embryonic cells of murine and human origin from pancreatic ducts that express insulin and respond to glucose stimulation.
Islet Cell Tumors
Overview and History
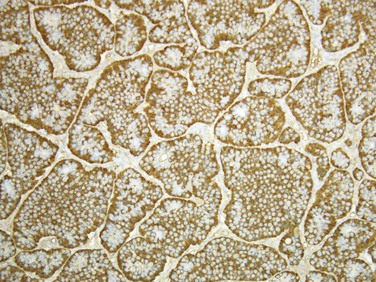
Endocrine tumors of the pancreas in the United States are rare, with an estimated incidence of 5 to 10 cases/1 million population annually. These tumors are 1000 to 2000 times more common in autopsy statistics, indicating that most are benign and nonfunctional. Endocrine tumors of the pancreas vary greatly in the mode of onset, severity of symptoms, location, functionality, and malignant potential.15 The incidence of malignancy in these tumors varies from approximately 10% in insulinomas to almost 100% in glucagonomas and somatostatinomas (see Table 40-1). On hematoxylin and eosin–stained sections, all pancreatic endocrine tumors look similar. Malignancy is determined by the presence or absence of metastases and immunostaining allows for the identification of the endocrine content of the cells (Fig. 40-4). Over time, they may vary significantly in secretion of hormone products and biologic aggressiveness. Although the tumor syndromes are classically attributed to pancreatic islet tumors, tumors are often found in extrapancreatic locations, such as the duodenum and peripancreatic soft tissue. Almost all insulinomas, glucagonomas, and VIPomas arise from the pancreas, whereas most gastrinomas occur in the duodenum. Stomatostatinomas are equally divided between the pancreas and proximal small bowel.
In 1908, Nichols described a pancreatic adenoma consisting of islet cell tissue. In 1935, Whipple and Frantz were the first to report an association between a clinical syndrome and an islet cell tumor. They described hyperinsulinism and the associated symptoms that became known as Whipple’s triad—the appearance during fasting of neuroglycopenic symptoms of hypoglycemia, low blood glucose (<45 mg/dL), and relief of symptoms by the administration of glucose. Over the next 25 years, additional syndromes associated with islet cell tumors were described. In 1942, Becker described a patient with severe dermatitis, anemia, and diabetes who also had an islet cell tumor; McGarvan later identified the cause of the syndrome as glucagon-secreting islet cell carcinoma of the pancreas. In 1955, Zollinger and Ellison described two patients with a fulminant peptic ulcer diathesis, acid hypersecretion, and non–beta islet cell tumors of the pancreas.16 It was later determined that the secretagogue was gastrin. The first description of watery diarrhea and hypokalemia related to an islet cell tumor was by Priest and Alexander in 1957. In 1958, Verner and Morrison described two patients who died from refractory watery diarrhea and hypokalemia and an associated islet cell tumor. Later, this syndrome was clearly defined when patients with this constellation of symptoms and islet cell tumors were found to have high circulating levels of VIP. The development and refinement of sensitive radioimmunoassay techniques in 1956 allowed for the detection of micromolar concentrations of circulating peptides and greatly contributed to our understanding of these syndromes.
Molecular Genetics of Islet Cell Tumors
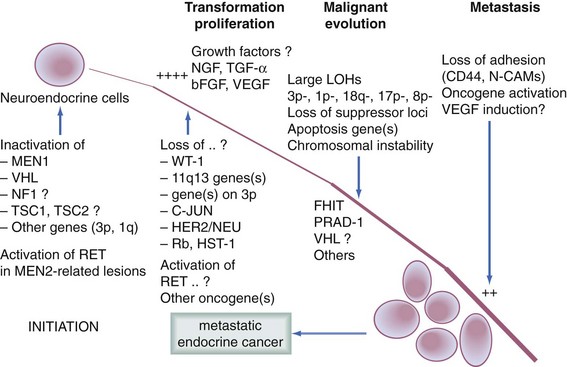
Similar to the adenoma-carcinoma sequence in colorectal cancer, tumorigenesis of islet cells and other neuroendocrine cells involves an accumulation of a number of genetic events, including activation of oncogenes and inactivation of tumor suppressor genes (Fig. 40-5). This progression is distinct from that of pancreatic adenocarcinoma. Mutations in the k-ras, p53, dpc4, myc, fos, jun, src, and retinoblastoma (mainly RB1) genes are not seen. Transcriptional silencing is believed to play a role in islet cell tumorigenesis. More than 90% of gastrinomas and nonfunctioning neuroendocrine tumors had homozygous deletions or epigenetic silencing by 5′ CpG island methylation.17 Loss of heterozygosity (LOH) at chromosome 11q is common in functional pancreatic endocrine tumors, whereas LOH at chromosome 6q is associated with the development of nonfunctional tumors.18 One third of patients with sporadic pancreatic endocrine tumors have allelic loss on chromosome loci 3p35, 3p27, and 11p13, suggesting that these loci encode tumor suppressor genes critical for the development of endocrine tumors. This allelic loss is associated with malignant clinical disease.19 More than 90% of these tumors demonstrate silencing of the tumor suppressor gene p16/MTS. Evers and colleagues20 have shown amplification of the proto-oncogene HER-2/neu, but not p53 or ras in gastrinomas. Others have reported an increase only in aggressive tumors.21 Studies of insulinomas have shown that the G protein Gs has threefold greater expression in insulinoma when compared with normal islet cells, suggesting that it may be involved in unregulated insulin secretion or tumorigenesis. Activation of the myc oncogene, TGF-α, and ras genes may be early genetic events in insulinoma tumorigenesis. Loss of the sex chromosome (X in women and Y in men) has been identified in pancreatic endocrine tumors and appears to be associated with an aggressive phenotype.
Although most pancreatic endocrine tumors occur sporadically, others can be associated with genetic syndromes. The most common genetic syndrome associated with pancreatic endocrine tumors is multiple endocrine neoplasia type 1 (MEN1). The syndrome is also characterized by pancreatic endocrine tumors, parathyroid hyperplasia, and pituitary adenomas. Pancreatic endocrine tumors occur in 30% to 80% of patients with MEN1 and are the most common cause of tumor-related death in MEN1 patients. MEN1 is caused by mutations or allelic deletions in the tumor suppressor gene, MENIN, on chromosome 11q13 and is inherited in an autosomal dominant fashion. Mutation or allelic deletion causes loss of tumor suppressor function and predisposes patients to neoplastic growth in the parathyroid, pituitary, and pancreatic endocrine tissue. Patients with MEN1-associated pancreatic endocrine tumors tend to be younger (30 to 40 years old), more likely to have malignant disease, and more likely to have multicentric disease than patients with sporadic tumors. Approximately 50% of patients with MEN1-associated neuroendocrine tumors will present with metastatic disease.22 Gastrinomas are the most common functional pancreatic endocrine tumors occurring in MEN1 patients (54% of functional MEN1-associated tumors). PPomas, which are not associated with a functional syndrome, occur most commonly in more than 80% of MEN1 cases.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


