Anderson-Fabry disease is a lysosomal storage disorder caused by α-galactosidase defects and progressive intracellular accumulation of globotriaosylceramide. The disease can be specifically treated with enzyme replacement therapy. Hemizygous men and heterozygous women can develop cardiac disease. Whereas men experience the most severe clinical phenotype, clinical presentation in women varies from asymptomatic to severely symptomatic. The characteristic cardiac phenotype is left ventricular hypertrophy mimicking sarcomeric hypertrophic cardiomyopathy or hypertensive heart disease. Early or prehypertrophy cardiac involvement may escape detection, unless electrocardiographic clues are present. The cardiac markers that raise suspicion of Anderson-Fabry disease include a short PR interval without a δ wave and a prolonged QRS interval, supraventricular and ventricular arrhythmias, and concentric left ventricular hypertrophy. Extracardiac features include renal failure, corneal deposits, and nervous, gastrointestinal, and cutaneous manifestations. Useful family data include cardiac and extracardiac traits in relatives and absence of male-to-male transmission. Symptoms are subtle, and the interval between the onset of symptoms and diagnosis may be as long as 20 years. As such, the diagnosis is typically late. Endomyocardial biopsy shows optically empty myocytes on light microscopy and dense osmiophilic bodies constituted of globotriaosylceramide on electron microscopy. Alpha-galactosidase A activity is reduced in hemizygous men but not in heterozygous women. Genetic testing is the gold standard for the diagnosis. In conclusion, a correct and timely diagnosis offers the possibility of disease-specific treatment that leads to sustained clinical benefits for cardiac and noncardiac signs and symptoms.
Anderson-Fabry disease (AFD) is an X-linked heritable disorder affecting glycosphingolipid catabolism, caused by deficiency or lack of the lysosomal enzyme α-galactosidase A activity. The incidence ranges from as high as 1 in 40,000 to 1 in 117,000 live male births. The enzyme defect leads to the accumulation of globotriaosylceramide and related glycosphingolipids in the plasma and cellular lysosomes. The disease is systemic, affecting cardiovascular, renal, cerebrovascular, neurologic, gastrointestinal, auditory, ophthalmologic, and dermatologic systems ( Table 1 ). The phenotypic manifestations are gender and age dependent. Hemizygous male patients demonstrate very low plasma and granulocyte enzyme activity and are diagnosed earlier than heterozygous women, who often do not demonstrate low enzyme activity and show later organ and tissue involvement. Whatever be the age and gender, symptoms usually appear up to 20 years before the diagnosis is made and are frequently unrecognized as potential clinical markers of the disease.
| Clinical Feature | Signs and Symptoms |
|---|---|
| Cardiac | LV hypertrophy, valve disease, short PR interval |
| Chest pain, dyspnea, fatigue, palpitations, arrhythmias, cerebrovascular accidents | |
| Cutaneous | Angiokeratoma, telangiectasia, hyperhidrosis, hypohidrosis, anhidrosis |
| Audiologic | Hearing impairment, sudden deafness, tinnitus, vertigo |
| Endocrine | Delayed puberty, growth delay |
| Ophthalmologic | Cornea verticillata, tortuous vessels, posterior subcapsular cataract, visual impairment |
| Gastrointestinal | Abdominal pain, constipation, diarrhea, nausea, vomiting |
| General feeling | Cold intolerance, heat intolerance, fever attacks |
| Musculoskeletal | Body pain, joint pain, joint stiffness, limb weakness, muscle pain |
| Neurologic | Peripheral: chronic pain, pain attacks, postural hypotension |
| Central: stroke, transient ischemic attacks | |
| Psychiatric | Depression |
| Renal/urinary | Hematuria, microalbuminuria, proteinuria, renal failure |
| Respiratory | Asthma |
| Vascular | Lymphedema |
AFD represents a treatable genetic disease involving the heart and often mimicking another genetic disorder, sarcomeric hypertrophic cardiomyopathy (HC), for which no specific treatment exists. It is therefore mandatory to make the correct differential diagnosis between sarcomeric HC and AFD-related left ventricular (LV) hypertrophy (1% of consecutive HC). If characteristic noncardiac traits of AFD are found associated with LV hypertrophy, the disease is suspected and specifically investigated. However, when noncardiac traits are absent or unrecognized, the diagnosis is usually sarcomeric HC. Until 2001, an incorrect diagnosis may have been inconsequential, but currently, the lack of a diagnosis would prevent the administration of enzyme replacement therapy (ERT); the European Medicines Agency and the United States Food and Drug Administration have approved ERT for the management of AFD. This review provides a clinically based workup to suspect and differentiate AFD HC from other types of HC.
Clinical Scenarios
In each of the following scenarios, the family history and clinical traits in probands and relatives are highly informative for suspecting AFD. A disease-oriented interview is the first low-cost step of the diagnostic workup.
Scenario 1
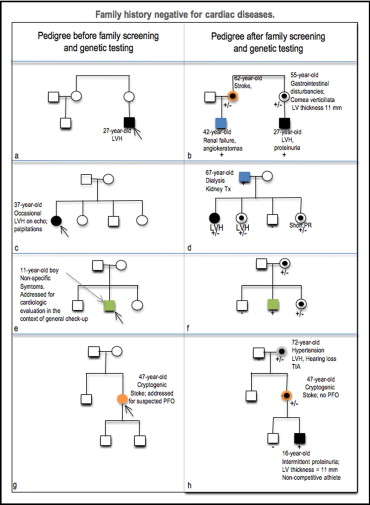
The proband is a young adult man with atypical symptoms and incidental detection of LV hypertrophy. In his family, there are no data suggesting familial HC ( Figure 1 ). Routine biochemical tests show mild proteinuria. Paternal history is negative; the mother reports vague gastrointestinal and/or ocular problems. Older male cousins are affected by renal insufficiency ( Figure 1 ). Cryptic strokes in the maternal lineage (5% to 6%), even in the presence of systemic, but not necessarily coincident, hypertension (18%), should be alerting, especially when associated with long-lasting nonspecific symptoms such as depression and pain attacks.
Scenario 2
The proband is a middle-aged woman referred for palpitations and incidental discovery of mild concentric LV hypertrophy. The cardiac family history is silent, but she describes a father with renal failure ( Figure 1 ). In this latter case, she may have affected sisters, but not affected brothers ( Figure 1 ). Unless specifically asked by the interviewing physicians, patients are unlikely to link the kidneys and the heart. In contrast, cardiologists who see patients with renal failure and LV hypertrophy may more frequently suspect other systemic diseases, such as amyloidosis. The patient should be further interviewed about symptoms that affect the quality of her life, such as acroparesthesia, hyperhidrosis or hypohidrosis, or abdominal pain. Ocular disturbances may not be apparent, and ophthalmologic evaluation searching for corneal deposits may help.
Scenario 3
The proband is a boy who presents for clinical evaluation because of tiredness, lethargy, and other nonspecific symptoms, such as episodes of abdominal and limb pain that starts in the hands and feet and can radiate proximally. His father is healthy ( Figure 1 ). For weight loss, the mother has been suspected of various unrelated disorders, including celiac disease that tested negative. Although it has been reported that children do not usually show myocardial involvement, electrocardiography shows a short PR interval even if echocardiographic results are normal. The family study may document genetically affected, asymptomatic older healthy sisters or symptomatic mutated brothers or healthy siblings ( Figure 1 ). The carrier mother shows normal electrocardiographic (ECG) and echocardiographic features.
Scenario 4
The proband is a woman who undergoes clinical evaluation for cryptogenic stroke with the specific request of excluding patent foramen ovale. Echocardiographic results are noncontributory. Her parents are apparently healthy ( Figure 1 ); the mother is hypertensive with echocardiographic evidence of LV hypertrophy and a history of transient ischemic episodes. Although cryptogenic stroke associated with LV hypertrophy may also occur in mitochondrial diseases as well as in other disorders, it recurs in women afflicted with AFD. The mother may also show cornea verticillata, gastrointestinal problems, and/or hearing loss ( Figure 1 ). The son of the proband shows a short PR interval and normal LV thickness on echocardiography.
Cardiologic Evaluation
Electrocardiography
Incidental findings of a short PR interval without a d wave (below the lower limit for age in children and <120 ms in adults ) in subjects who undergo electrocardiography for cardiac or noncardiac reasons (presurgery evaluation, sport suitability, etc.) should be considered with caution as early potential markers of a myocardial storage disease ( Figure 2 ). A short PR interval is seen in 21% to 40% of adult patients with AFD and in about 28% of pediatric patients. The PR interval may change with age and/or the grade of the disease. It may be (1) short in affected men (up to 40%), (2) short in children without any other signs of the disease, (3) normal in children with echocardiographic evidence of early or mild LV hypertrophy, and (4) prolonged in the advanced phases of the disease until high-grade atrioventricular block.
In adults, ECG signs of LV hypertrophy are present in up to 61% of men and 18% of women. QRS duration has been reported up to 160 to 200 ms, and the 12-lead amplitude-duration product is reported to show the best correlation with LV mass. The product is calculated as the sum of the total QRS amplitudes in the 12 ECG leads (mm) multiplied by the QRS duration, choosing the longest QRS duration in lead I, II, or III. The Sokolow-Lyon product shows a good relation with the LV mass estimated by echocardiography and is calculated from the Sokolow-Lyon index (the sum of SV 1 and RV 5 voltages) when it is multiplied by the longest QRS duration in lead I, II, or III ; a cut-off value >297 (sensitivity >80%) has 46% specificity. The Cornell product shows similar results.
Ambulatory ECG monitoring
Cardiac arrhythmias in affected children are about 3%, a percentage that can be considered quite high compared to the normal population (up to 22.5 per 100,000 patients aged <18 years presenting to emergency departments ). The most common arrhythmia is sinus bradycardia, followed by ectopic atrial rhythms.
Although sudden arrhythmic death is rare (0.1%), arrhythmias are common in adult male patients. In the late phase of the disease, almost all affected adult men show nonsustained ventricular tachycardia on Holter ECG monitoring and nonspecific intraventricular conduction disturbances, with QRS duration up to 160 to 200 ms. Arrhythmias may recur independently of the presence of LV hypertrophy. About 6% of patients undergo pacemaker or defibrillator implantation.
Heart rate variability
Heart rate variability correlates with autonomic dysfunction and therefore with dyshidrosis or hypohidrosis. In women aged <18 years, heart rate variability is normal, while in men of the same age, it is significantly lower than in normal subjects matched for age.
Echocardiography
In adult patients with cardiac disease, the left ventricle shows mild to moderate concentric hypertrophy (14 to 20 mm). LV mass is increased in up to 76% of patients and in almost all women aged >45 years ( Figure 2 ). In consecutive series of patients with unexplained LV hypertrophy, 1% to 4% are affected by genetically proved AFD. LV mass is increased when echocardiography shows LV hypertrophy, and it is >75th percentile of the normal population when echocardiography is normal. Up to 50% of patients with LV hypertrophy also show selective and focal hypokinesia of the basal LV posterior wall and a corresponding thinning of the wall (4 to 7 mm).
The cardiac valves are mildly involved in the disease and show thickening of the aortic and mitral leaflets ( Figure 2 ) in 20% of children and about 50% of adults (57% mitral valve, 47% aortic valve ). The resulting mild “stiff” mitral prolapse should be distinguished from myxomatous “floppy” valve prolapse. M-mode analysis of mitral leaflet movement may contribute to the differentiation of the 2 conditions ( Figure 2 ).
In Anderson-Fabry cardiomyopathy, systolic and diastolic function is impaired. In children with AFD, systolic and diastolic cardiac function is normal, independent of evidence of cardiac hypertrophy. In adolescents, initial LV remodeling or hypertrophy is present in 50% of female patients and 62% of male patients. Progressive systolic and diastolic dysfunction becomes evident with increasing age. Early reductions of systolic and diastolic tissue Doppler imaging indexes are detected before the onset of myocardial hypertrophy, the longitudinal performance being impaired before the radial one. In echocardiographic series of patients diagnosed with AFD compared to normal subjects, tissue Doppler imaging values do not overlap with those of healthy subjects. Lateral and septal Sa and Ea <10 cm/s have near 100% sensitivity and specificity in identifying mutation-positive subjects without LV hypertrophy. Tissue Doppler imaging also constitutes an accurate and noninvasive tool for the assessment of cardiac improvement during ERT.
Electrophysiologic evaluation
The few existing data draw attention to the short PR interval. The changes in PR interval are thought to be due to glycosphingolipid accumulation in the myocardial fibers and conduction system. The most relevant contribution is the exclusion of accessory pathways causing atrioventricular shortening. The short PR interval seems to be a reversible characteristic of AFD, as it can normalize after ERT. This last consideration reinforces the hypothesis that glycosphingolipid accumulation is the most probable cause of the accelerated atrioventricular conduction.
Cardiac magnetic resonance
Cardiac magnetic resonance provides information on LV hypertrophy and may detect delayed enhancement (DE) that is related to the presence of focal scarring. The DE distribution characteristically spares the subendocardium. The prevalence of DE is similar in male and female patients; the extent is greater in men, where it seems to be related to the severity of LV hypertrophy. In 92% of DE-positive patients, the location is in the basal posteroinferior LV wall. This feature correlates with the echocardiographic finding of selective thinning of the basal inferoposterior wall compared to other myocardial segments. DE may be the substrate for electrical reentry and sudden arrhythmic death. The focal fibrosis may reflect inhomogenous LV wall stress or relative myocardial ischemia even in the presence of normal epicardial coronary arteries.
Endomyocardial biopsy
Endomyocardial biopsy plays a major role in the diagnosis, as (1) it confirms that echocardiographic LV hypertrophy is the result of intramyocyte accumulation of glycosphingolipids (disease-related myocardial damage), and (2) it may show accumulation of globotriaosylceramide in the myocardium of patients with borderline LV hypertrophy, offering proof of myocardial involvement.
Light microscopy shows optically empty myocytes ( Figure 3 ). Semithin sections show intramyocyte accumulation of osmiophilic bodies ( Figure 3 ), and ultrastructural study documents the osmiophilic lamellar bodies typical of AFD ( Figure 3 ). Because the myocardial toxicity in patients treated long term with chloroquine or with amiodarone constitutes true pathologic phenocopies of AFD, the pathologic report is consistent with AFD, but the final diagnosis is formulated when the disease is genetically confirmed.
Cardiologic Evaluation
Electrocardiography
Incidental findings of a short PR interval without a d wave (below the lower limit for age in children and <120 ms in adults ) in subjects who undergo electrocardiography for cardiac or noncardiac reasons (presurgery evaluation, sport suitability, etc.) should be considered with caution as early potential markers of a myocardial storage disease ( Figure 2 ). A short PR interval is seen in 21% to 40% of adult patients with AFD and in about 28% of pediatric patients. The PR interval may change with age and/or the grade of the disease. It may be (1) short in affected men (up to 40%), (2) short in children without any other signs of the disease, (3) normal in children with echocardiographic evidence of early or mild LV hypertrophy, and (4) prolonged in the advanced phases of the disease until high-grade atrioventricular block.



