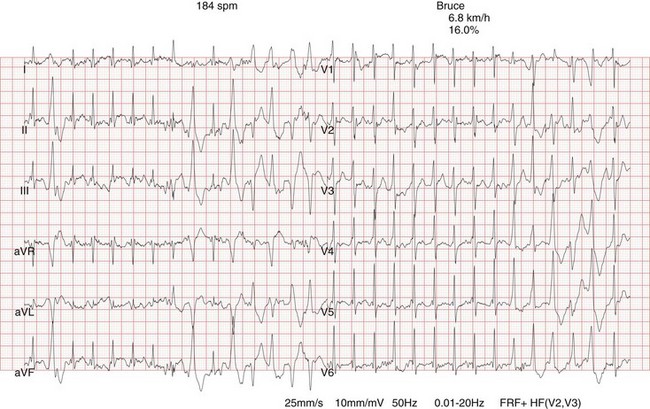
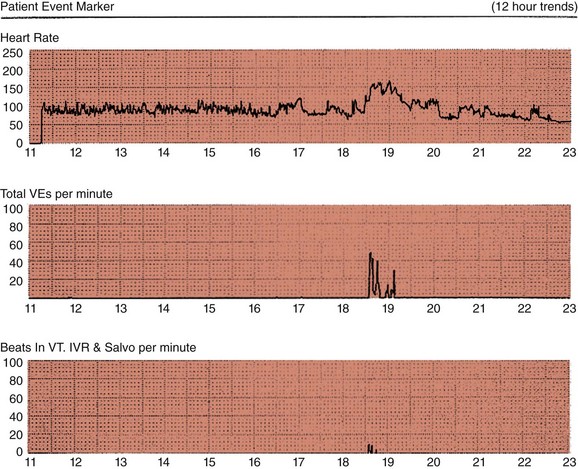
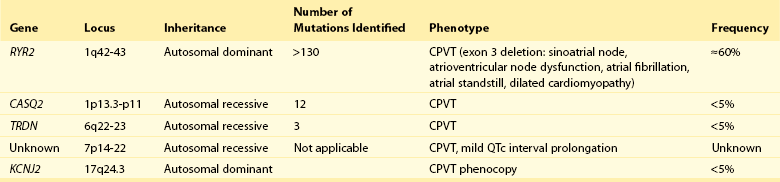
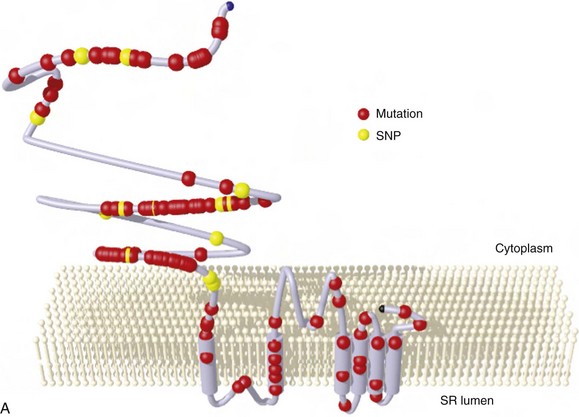
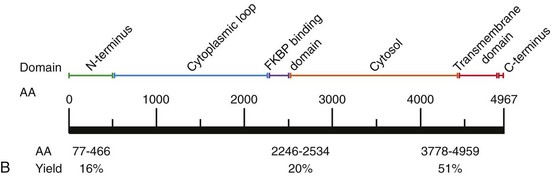
88 Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited arrhythmia syndrome that is characterized by adrenergically mediated polymorphic ventricular tachyarrhythmias in patients with no structural heart disease. The prevalence of CPVT in the general population is unknown but is estimated to be 1 in 10,000. Although the clinical course is diverse, mortality in severely affected untreated CPVT patients is up to 50% before the age of 20.1 Accordingly, CPVT is a significant cause of sudden infant death syndrome (SIDS; see Chapter 98) and autopsy-negative sudden unexplained death in the young.2 The first patients with the clinical characteristics of CPVT were described in 1960 by Berg, who reported three young sisters with cardiac events and polymorphic ventricular arrhythmias without structural heart disease, of whom one died suddenly.3 In 1975, Reid et al. described another typical CPVT patient: a 6-year-old girl with bidirectional ventricular tachycardia precipitated by exercise or emotional stress.4 Thereafter, the Paris group (Lariboisière Hospital) of Philippe Coumel published two important clinical articles in 19785 and 1995,1 including 4 and 21 patients respectively, resulting in the definitive recognition of CPVT as a distinct primary arrhythmia syndrome (instead of a subtype of the long QT syndrome). The genetic background of CPVT was first discovered in 2001: Mutations in the cardiac ryanodine receptor gene (RYR2) were found to underlie most cases of CPVT,6 while mutations in cardiac calsequestrin (CASQ2) were identified in rare, autosomal recessively inherited CPVT cases.7 Patients with a possible clinical diagnosis of CPVT are those with physical or emotional stress-related polymorphic ventricular ectopy but with no or no reproducible polymorphic ventricular arrhythmias. In these cases genetic testing is critical for a definitive diagnosis of CPVT. In genotype-negative cases with a possible CPVT phenotype, it may be challenging to distinguish CPVT from other, similar conditions (see Differential Diagnosis), and it may not be possible to make a definitive diagnosis. Today, however, numerous patients and families with a different clinical presentation and a more benign course have been recognized.8 On the contrary, RYR2 mutations have been identified in victims of SIDS, suggesting a wide range of phenotype severity among RYR2 mutation carriers. Most CPVT patients have an unremarkable 12-lead resting electrocardiogram, including a normal QTc interval. However, sinus bradycardia and prominent U waves on the resting electrocardiogram have been associated with CPVT (see Supraventricular Disease Manifestations). The golden standard for the diagnosis of CPVT is provocative testing, preferably performed through exercise. Typically, a gradual increase in ventricular arrhythmia burden and complexity is observed, starting with isolated ventricular premature beats (VPBs) at a heart rate of approximately 110 to 130 beats per minute. In the absence of important therapeutic modifications, the ventricular arrhythmia threshold heart rate is accurately reproducible in an individual patient. VPBs are late-coupled with a coupling interval of approximately 400 milliseconds. Left bundle branch block inferior axis and right bundle branch block superior axis morphologies have consistently been shown to be predominant in CPVT, and VPB morphologies are usually reproducible in an individual patient. Further exercise causes the number of isolated VPBs to increase to match the number of bigeminal VPBs, and eventually, polymorphic couplets or nonsustained ventricular tachycardia, including bidirectional ventricular tachycardia, may be induced (Figure 88-1). In very rare cases, this may further escalate to polymorphic sustained ventricular tachycardia or ventricular fibrillation. When exercise testing is terminated, the ventricular arrhythmias rapidly recede in most patients. Figure 88-1 Electrocardiographic strip showing the typical bidirectional ventricular tachycardia in a patient with catecholaminergic polymorphic ventricular tachycardia during exercise testing. Holter monitoring, during which a patient should be encouraged to perform exercise, can be used as an alternative test in selected cases, although its sensitivity is thought to be low. For example, it may be useful in young children or in other patients who are unable to perform an adequate exercise test, in patients suspected of having emotion-related rather than exercise-related ventricular arrhythmias, and in patients who report possible CPVT-related symptoms but have an unremarkable exercise test (Figure 88-2). Figure 88-2 Holter monitoring summary of a patient with catecholaminergic polymorphic ventricular tachycardia showing the occurrence of ventricular premature beats (middle panel) and ventricular tachycardia (lower panel) when heart rate is increased (upper panel). In recent years the use of adrenaline infusion (initiated at a dose of 0.05 µg/kg/min and then titrated at 5-minute intervals to a maximum dose of 0.2 µg/kg/min) in CPVT has been advocated. It is interesting to note that adrenaline infusion–provoked ventricular arrhythmias consistent with CPVT have been reported in patients who did not have any ventricular arrhythmia during exercise testing or Holter monitoring. Whether these patients represent “true” CPVT based on a similar pathophysiological mechanism as that seen in classic CPVT patients remains to be determined. A recent study comparing the diagnostic value of adrenaline infusion and exercise testing in 36 CPVT patients (including 25 RYR2 mutation carriers and 11 genotype-negative patients) and in 45 unaffected relatives showed low sensitivity of adrenaline infusion, probably because the maximum heart rate achieved upon adrenaline challenge was markedly lower as compared with that seen during exercise testing.9 Among 25 CPVT patients with a positive exercise test, only 7 had a positive adrenaline test (sensitivity of 28%). The specificity of adrenaline infusion in the entire study population was 98%.9 Electrophysiological studies generally have no role in diagnosing CPVT because ventricular arrhythmias cannot be triggered other than by adrenergic stimulation. However, in selected cases, they may be useful: A recent article reported on prominent postpacing changes in the QT interval among mutation carriers from a family with the M4109R RYR2 mutation.10 In an index patient who presents with a possible CPVT phenotype, cardiac imaging is mandatory to exclude other causes of exercise-induced polymorphic tachyarrhythmias (see Differential Diagnosis). Cardiac imaging is, by definition, unremarkable in CPVT patients. However, some exceptions have been reported. Mutations in RYR2 have been identified in patients with fibrofatty myocardial replacement in the right ventricle, mimicking arrhythmogenic cardiomyopathy, and intracellular calcium deposits.11 Moreover, members of two separate families with a large genomic deletion in RYR2, involving exon 3, showed sinoatrial node and atrioventricular node dysfunction, atrial fibrillation, atrial standstill, and left ventricular dysfunction and dilatation, in addition to the classic CPVT phenotype.12 The most common supraventricular manifestation of CPVT is sinus bradycardia and/or sinus node dysfunction. In one study, RYR2 mutation carriers had a lower average resting heart rate as compared with their non–mutation-carrying relatives.13 Heart rates tended to be lowest in males and in RYR2 mutation carriers with a CPVT phenotype. The latest data on RYR2 mutation–carrying relatives, including 116 relatives from 15 families who were identified by cascade screening of the RYR2 mutation causing CPVT in the proband, show that sinus bradycardia was observed in 19%.14 Other supraventricular dysrhythmias were present in 16% and mainly included intermittent ectopic atrial rhythm identified by Holter monitoring. In one study, in which an electrophysiological study was performed in eight CPVT patients, evidence of sinus node dysfunction was demonstrated in four.15 Exercise-induced supraventricular tachyarrhythmias have been reported in CPVT patients but are not commonly observed.14 Although the familial nature of CPVT was noted in the first reports, critical steps in revealing the genetic basis of CPVT have been taken in the past decade (Table 88-1). The pathophysiological mechanisms through which these genetic determinants of CPVT lead to the clinical phenotype are discussed in Chapter 53. Table 88-1 Genetics of Catecholaminergic Polymorphic Ventricular Tachycardia CPVT, Catecholaminergic polymorphic ventricular tachycardia. In 1999 the CPVT phenotype was linked to a disease locus on chromosome 1q42-q43, and an autosomal dominant inheritance pattern of this CPVT form was suggested. The disease-causing gene residing on this locus—the cardiac ryanodine receptor gene (RYR2)—was identified in 2001. RYR2 governs the release of calcium from the sarcoplasmic reticulum, which initiates cardiac muscle contraction. More than 130 unique and mostly missense mutations in RYR2 have been identified.16 Approximately 20% of RYR2 mutations are de novo in origin. Mutations in the RYR2 cluster have been reported in three hotspots: the N-terminal domain (codons 44 to 466; ≈16% of mutations), the central domain (codons 2246 to 2534; ≈20% of mutations), and the C-terminal channel–forming domain (codons 3778 to 4959; ≈50% of mutations) (Figure 88-3). Nowadays, but probably not for long because of advanced technological possibilities enabling more extensive molecular genetic screening, most laboratories use this information by applying a tiered targeting strategy for RYR2 mutational analyses, starting with the exons in which most mutations have been identified.16 Figure 88-3 A, RYR2 channel topology and localization of mutations and single-nucleotide polymorphisms (based on Medeiros-Domingo et al.16). SNP, Single-nucleotide polymorphism; SR, sarcoplasmic reticulum. B, Proportion of RyR2 mutations identified per domain (indicated by the amino acid number estimated for each domain) (based on Medeiros-Domingo et al.16). AA,
VTs in Catecholaminergic Cardiomyopathy (Catecholaminergic Polymorphic Ventricular Tachycardia)
Historical Background
Clinical Manifestations
Definition
Clinical Presentation
Cardiologic Examination
Supraventricular Disease Manifestations
Genetic Basis
Cardiac Ryanodine Receptor Gene (CPVT 1)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


VTs in Catecholaminergic Cardiomyopathy (Catecholaminergic Polymorphic Ventricular Tachycardia)
